

 PDF
PDF ePub
ePub Citation
Citation Print
Print


The most common cause of cardioembolic stroke is non-valvular atrial fibrillation. Other causes include cardiomyopathy, which occasionally produces mural thrombosis. Cardiac involvement of amyloidosis is not uncommon in systemic amyloidosis associated with multiple myeloma, and is one of the causes of infiltrative cardiomyopathy in adults.1 However, cerebral embolism in relation to cardiac amyloidosis has rarely been reported in the neurology literature. We report two patients who had embolic infarcts associated with cardiac amyloidosis. The diagnosis was based on the biopsy of myocardium (Patient 1) and the kidney biopsy and echocardiographic results (Patient 2).
CASE REPORT
Patient 1
A 29-year-old woman with known cardiac amyloidosis suddenly developed three episodes of transient left hemiparesis. The patient had first been seen at our hospital three months earlier because of dyspnea on exertion (NYHA class III). Electrocardiogram (ECG) revealed sinus rhythm with low voltage QRS and Q waves in leads V2-3. Radiographs of the chest showed cardiomegaly with bilateral pleural effusion. Transthoracic echocardiography (TTE) showed thickened left ventricular (LV) wall with high left ventricular end-diastolic pressure (LVEDP) and inferior vena cava (IVC) plethora that reflected the high pressure of right atrium (ejection fraction=40%). Both atria were enlarged and transmitral doppler flow revealed typical restrictive pattern. These findings were consistent with restrictive cardiomyopathy. Microscopical examination of a specimen from endomyocardial biopsy, stained with Congo red, showed amyloid deposition (Fig. 1). Serum and urine protein electrophoresis showed no abnormal zone of restriction with monoclonal antibodies. Bone marrow, rectum and abdominal fat aspiration biopsies showed no abnormality. Nerve conduction studies revealed no evidence of neuropathy. Based on these findings, a diagnosis of cardiac amyloidosis was made. There was no evidence of systemic involvement nor family history of amyloidosis. Torasemide, amiloride and spironolactone were prescribed and heart transplantation was under consideration.
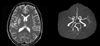
She had no history of hypertension and diabetes. She had never smoked and did not drink alcohol. On examination, cardiac murmur and cervical bruit were not heard. ECG showed sinus rhythm with occasional premature ventricular complexes, low voltage QRS and Q waves in leads V2-3. Neurologic examinations revealed no abnormality. Diffusion-weighted magnetic resonance imaging (MRI) 6 hours after the onset showed a high signal lesion in the right basal ganglia and corona radiata. MR angiography (MRA) showed near total occlusion (or severe stenosis) of the distal M1 and proximal M2 portion of the right middle cerebral artery (MCA) (Fig. 2). On the sixth hospital day, follow-up MRI and MRA revealed no interval changes. TTE showed thickened LV wall with granular sparkling and both atrial filling pressure (ejection fraction=28%). These findings were compatible with cardiac amyloidosis and aggravated LV dysfunction compared to previous study three months before. There was mild spontaneous echo contrast (SEC) without thrombi on transesophageal echocardiography (TEE). The patient was treated with heparin and later with warfarin. She remained asymptomatic.
Patient 2
A 73-year-old woman suddenly developed global aphasia, right hemiplegia and visual disturbances. She had hypertension and had been treated with medications during the last 5 years. The patient had first been seen at our hospital one year earlier because of dyspnea on exertion. ECG revealed ST elevation in leads V2-3 and inverted T waves in leads V2-5. Echocardiography showed concentric left ventricular hypertrophy (LVH) with increased LVEDP.
Two months before admission, the patient visited our hospital because of pretibial pitting edema. Laboratory test results were consistent with a nephrotic syndrome. (proteinuria; 4.2 g/day, hypoalbuminemia; 2.7 g/dL, hypercholesterolemia; 294 mg/dL) Serum protein electrophoresis showed an abnormal zone of restriction in the gamma region, identified as an IgG M component with lambda light-chain restriction on immunofixation. Urine protein electrophoresis revealed an abnormal zone of restriction in the gamma region, identified as Bence-Jones protein and IgG lambda M component, respectively, on immunofixation. Kidney biopsy, stained with Congo red, demonstrated amyloid deposition and apple-green birefrigence with polarized light (Fig. 3). Bone marrow biopsy of the left iliac crest showed moderate plasmocytosis (6.2%). Skeletal survey showed diffuse osteopenia of the bony structures, but no lytic foci on the skull. Base on these findings, a diagnosis of multiple myeloma with amyloidosis was made.2 Further history taking revealed that there was no family history of amyloidosis. Thalidomide and dexamethasone was prescribed.
The patient had never smoked and did not drink alcohol. On examination, cardiac murmur and cervical bruit were not heard. Radiographs of the chest showed cardiomegaly with pulmonary congestion. ECG revealed a sinus rhythm with multiple atrial premature complexes. On neurologic examination, she was alert. Her eye balls were deviated to the left. There were global aphasia, right hemiparesis (I/V), right facial paresis and left homonymous hemianopia. Brain computed tomography (CT) 2 hours after the onset showed a low density lesion in the right posterior cerebral artery (PCA) territory, but no evidence of early ischemic change in the left MCA territory. Brain CT angiography showed missing of some of the left MCA cortical branches, suggesting distal MCA occlusion. After the intravenous infusion of tPA (0.6 mg/kg), transfemoral cerebral angiography showed the recanalization of most of the left MCA branches except for an occlusion of one of the frontal branches, probably due to emboli migration.
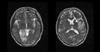
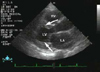
On the fifth hospital day, follow-up MRI revealed acute infarcts in the right PCA and left MCA territories (Fig. 4). MRA showed no abnormality of intracranial arteries, suggesting complete recanalization of the vessel. TTE showed symmetrical thickening of the LV wall with high LVEDP and IVC plethora that reflected the high pressure of the right atrium (ejection fraction=50%). Both atria were enlarged and transmitral doppler flow revealed typical restrictive pattern. These findings were consistent with restrictive cardiomyopathy, and the cardiac involvement of amyloidosis was strongly suspected (Fig. 5). There were left atrial appendage thrombi (17*16 mm) with vigorous biatrial SEC on TEE. Cardiac biopsy was not performed because this procedure was considered too invasive for her condition.
Motor nerve studies revealed low amplitude of compound muscle action potentials in left ulnar and left peroneal nerves and normal velocity in all tested motor nerves. Sensory nerve action potentials could not be detected in the legs. These findings were consistent with the presence of a sensory dominant polyneuropathy.
The patient was treated with low molecular weight heparin and later with warfarin. Her neurological condition improved and there were mild mixed aphasia and no hemiparesis at one week after the onset. One month later, she did not have any neurological deficits.
DISCUSSION
Patient 2 had cerebral infarcts in the territories of right PCA and left MCA. There was an early MCA recanalization and no remarkable atherosclerotic changes in MRA. Patient 1 had a cerebral infarct in the right basal ganglia and corona radiata, and near total occlusion of the MCA. Although there was no MCA recanalization, this young lady had neither vascular risk factors nor remarkable atherosclerotic changes in MRA. These findings suggest that cardiac embolization was the etiology of their stroke. In Patient 1, cardiac amyloidosis was diagnosed by the biopsy of cardiac muscles. In Patient 2, the diagnosis was supported by characteristic echocardiographic findings and amyloid deposits in the kidney.
The echocardiographicy findings in our patients included symmetrical thickening of the left ventricle wall with high LVEDP and IVC plethora. Both atria were enlarged and transmitral doppler flow revealed severe restrictive pattern. Although characteristic diffuse hyperrefractile "granular sparkling" appearance.3,4 was not observed in Patient 2, these findings are consistent with restrictive cardiomyopathy secondary to cardiac amyloidosis. Because Patient 2 had hypertension, one may argue that the cardiomyopathy was caused by long standing hypertension. However, this is highly unlikely. First, ECG demonstrated low voltage signals, despite increased thickness of LV walls on TTE. Second, typical restrictive pattern of mitral inflow suggested high LVEDP. Third, high right atrial pressure was associated with IVC plethora, which was not consistent with hypertensive cardiomyopathy. Moreover, the rapid progression of the echocardiographic findings one year after the first examination (LV posterior wall thickening, 17mm vs. 14mm; interventricular septum thickening, 17mm vs. 14mm; left atrial size, 48mm vs. 41mm; increased myocardial echogenecity for one year) was not consistent with usual hypertensive cardiomyopathy, either.
Other causes of restrictive cardiomyopathies such as sarcoidosis, endomyocardial fibrosis, radiation, toxin5 were ruled out by the history, physical examination and laboratory findings. Therefore, it is highly likely that Patient 2 had cardiac involvement of amyloidosis. Although definitive diagnosis should be based on biopsy findings, this invasive procedure was not performed in Patient 2 who developed acute stroke. On the other hand, Patient 1 was diagnosed as having cardiac amyloidosis by myocardial biopsy.
The mechanisms of embolization in amyloid cardiomyopathy are multifactorial. First, mural thrombosis may develop due to blood stasis and intracavitary turbulence. Deposition of amyloid fibrils results in reduced ventricular compliance with impairment of relaxation and eventually contraction.6-8 Second, amyloid may infiltrate into the intima of coronary arteries, which may cause myocardial ischemia, endocardial damage, mural thrombosis and subsequent cerebral embolism.6 Third, arrhythmia may develop in these patients. Atrial fibrillation occurs in about 20% of amyloid cardiomyopathy due probably to left atrial dilatation and failure with increased wall stress.9,10 The most likely mechanism for embolization in Patient 2 was the first one, since she had mural thrombus without evidence of myocardial ischemia or atrial fibrillation. In Patient 1, neither thrombi nor atrial fibrillation was observed. She did not belong to one of the above.
Although only SEC was observed on TEE of Patient 1, prophylactic anticoagulation was administered because of the severe LV dysfunction and global LV hypokinesia. In Patient 2, anticoagulation was administered because left atrial appendage thrombi were observed on TEE. However, anticoagulants should be used cautiously in these patients as systemic amyloidosis may be associated with coagulation abnormalities predisposing to bleeding.5,12 Although Patient 1 remained asymptomatic, and Patient 2 improved after thrombolysis and anticoagluation, the ultimate prognosis of these patients is considered poor. According to previous literatures, symptomatic heart involvement is the most powerful prognostic factor, associated with a median survival of only 6 months.3,11


 XML Download
XML Download