

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Osmotic demyelination syndrome (ODS) refers to central pontine myelinolysis (CPM) and extrapontine myelinolysis (EPM) following a marked change in osmolarity, usually due to the rapid correction of hyponatremia. These extrapontine lesions of ODS are most frequent in the neostriatum, lateral thalamus, internal capsule, midbrain, and cerebellum. However, cortical laminar necrosis (CLN), as shown by MR, has rarely been reported in ODS.1,2,3 We report a patient who developed ODS accompanying CLN, which was shown by MRI and MR spectroscopy (MRS).
CASE REPORT
A 32-year-old female patient was admitted to a local hospital after a car accident. A neurologic examination at admission was normal. Brain CT showed mild traumatic subdural and subarachnoid hemorrhage. The patient was treated with mannitol to decrease intracranial pressure. During mannitolization she became somnolent, and her serum sodium level was 97 mEq/ml. After 2 days, the sodium level had risen to 138 mEq/ml and her mental status was improved. On the 3rd day, the patient became drowsy and had a brief episode of generalized tonic-clonic seizure. No marked hypoxemia or hypotension was identified with closed hemodynamic monitoring in the intensive care unit. The patient gradually deteriorated and became comatose. MRI showed no significant abnormalities.
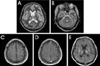
Four days after the correction of hyponatremia, the patient was transferred to our hospital. She was semicomatose with decerebrate posturing. Brainstem reflexes were all preserved. Babinski signs were present bilaterally. Two weeks after the correction of hyponatremia there were high signal intensities in the pons and bilateral deep gray nuclei on T2-weighted MRI images (Fig. 1-A and 1-B). T1-weighted images demonstrated multifocal curvilinear hyperintensities along the cerebral cortices (Fig. 1-C) with diffuse gyriform enhancement alongside most of the cerebral cortices and with a symmetrical enhancement of both external capsules, globus pallidi, and central thalami (Fig. 1-D and 1-E). Proton MRS was carried out using a 1.5-T system with a standard quadrature head coil. A stimulated echo acquisition mode sequence was used with a repetition time of 3 seconds, echo time of 30 milliseconds, and voxel volume of 8 ml placed at the parietal parasagittal cortex. This revealed an abnormal increase in lipid/lactate complex (at 0.8-1.6 ppm) and a decrease in N-acetyl aspartate (NAA) (at 2.0 ppm).
The patient became stable without a significant change in her level of consciousness. At a 1-year follow-up after admission, she was still in a vegetative state and did not respond to painful stimuli. The patient continued to show no awareness even when in an awakened state. However, her brainstem functions were well preserved.
DISCUSSION
Hypoxia/ischemia is the most probable cause of CLN evident on MRI, but neither hypoxia nor hypotension was identified in our patient. A single attack of generalized seizure was not thought to be related to a significant hypoxic event, which was supported by the gradual deterioration of the patient's mental status after recovery from a brief seizure.
Cortical laminar changes had developed in this patient after a rapid correction of hyponatremia. The patient also exhibited well-described MRI characteristics of ODS, including CPM, and no hippocampal lesions - which are commonly involved in hypoxic/ischemic injury - were found. A severe case of cortical lesions in ODS was previously found to include neuronal loss in addition to demyelination and gliosis.4 In the case reported here, CLN was believed to occur within the spectrum of ODS.
A patient with Sheehan syndrome suffering from hyponatremia and cardiac failure exhibited CPM and CLN on MRI.1 Another patient, who suffered from acute intermittent porphyria, also had severe hyponatremia and convulsions, and MRI showed CPM, EPM, and CLN.2 That patient was assumed to have arterial disease from acute intermittent porphyria in addition to hyponatremia and generalized convulsion. In contrast to the case reported here, both of these previous patients experienced a definite hypoxic/ischemic event resulting in CLN. One ODS case similar to ours who experienced decreased consciousness and one seizure attack showed cortical abnormality on MR, leading to two suggested underlying mechanisms: (i) pure ODS or (ii) ODS with minor hypoxic/ischemic injury.3
The brain is known to be more vulnerable to hypoxic ischemic injury in metabolic encephalopathy.5,6 Another possible explanation for the development of CLN in our patient is the vulnerability to hypoxic ischemic injury in ODS - even one episode of generalized seizure attack might contribute to ischemic injury in this state.
Cortical MRI abnormalities observed in patients with anoxia or ischemia pathologically represent CLN and invariably are predictive of a poor prognosis. In the case of ODS, however, underlying pathological changes are predominantly demyelinative with the degree of neuronal loss depending on the severity of the condition.2,4 The prognosis of cortical lesions in ODS is thought to be related to the degree of neuronal loss. Although the pathogenesis of ODS is still unknown, this condition may result from a breakdown of blood-brain barrier, leading to vasogenic edema, and subsequently progressing into demyelination accompanied by a variable degree of neuronal loss.4,7,8 Gadolinium-enhancing cortical MRI abnormalities in ODS indicate the breakdown of blood-brain barrier but not neuronal loss. In the present case we used MRS to determine the contribution of different pathogenesis such as ischemia and demyelination. NAA is a marker of a healthy neuron whose reduction indicates neuronal disease or loss, including infarction after stroke. Another remarkable finding in our case was the increase of broad peak intensities at 0.8-1.6 ppm, which were assigned to the lipid/lactate complex. The NAA peak decreased as the lipid/lactate complex increased. However, the interpretation was limited by the echo time being too short to differentiate between lipid and lactate. MRS revealed greater ischemic injury than demyelination in the cortical lesion. Moreover, the patient remained in a vegetative state, suggesting a poor prognosis.
In conclusion, CLN revealed by MR can occur with CPM and EPM, although the early detection of a cortical lesion is difficult. It is unclear whether pure hypoxia/ischemia coexisting with ODS or severe cortical neuronal loss is the main pathology of demyelination. The present case indicates that even a mild hypoxic/ischemic event should be avoided in the management of patients with osmotic disequilibrium.
 XML Download
XML Download