

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Calcinosis cutis is the deposition of insoluble calcium salts in the skin and subcutaneous tissue. Calcinosis cutis is divided into five subtypes: dystrophic calcification, metastatic calcification, idiopathic calcification, iatrogenic calcification, and calciphylaxis1. Dystrophic calcinosis is the most common subtype of calcinosis cutis which is characterized by normal calcium and phosphate levels in serum1. Dystrophic calcification appears as a result of local tissue damage and is most frequently observed in underlying autoimmune connective tissue disease (ACTD). Dermatomyositis (DM)2 and systemic sclerosis are the most common ACTDs which can be seen in calcinosis cutis3.
Calcinosis cutis in DM is frequently observed, especially in the juvenile-onset DM (JDM). However, calcinosis in juvenile-onset amyopathic dermatomyositis (amyopathic JDM) is rare.
CASE REPORT
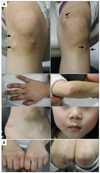
A 6-year-old female presented with 0.5 to 2 cm-sized multiple hard subcutaneous masses on both knees for 3 years. The masses were painless and growing slowly. The patient also presented with multiple erythematous scaly patches and plaques on bilateral knuckles, elbows, ankles and cheeks from the first year of life (Fig. 1A). Her mother had similar skin lesions which were erythematous scaly patches on the knuckles, knees and elbows, since her childhood (Fig. 1B). Both patient and her mother had no history of muscle weakness, photosensitivity or arthralgia.
Skin biopsies were performed from two sites from the patient. When skin biopsy was performed from a left knee nodule, liquid chalky discharge which is called "milk of calcium" was observed. The biopsy results were consistent with calcinosis cutis which showed tumorous deposition with bursa-like structure lined by calcifying front (Fig. 2A). Another biopsy from erythematous scaly plaque on the right elbow showed focal hydropic degeneration of basal keratinocytes and lichenoid inflammatory cell infiltration (Fig. 2B). Skin biopsy was also performed from her mother. Histopathologic findings from erythematous patch on mother's knee were mild basal vacuolar change and perivascular lymphohistiocytic cell infiltration (Fig. 2C).
Laboratory findings showed normal range of serum phosphorus (4.5 mg/dl), calcium (9.3 mg/dl), 1,25-dihydroxy-vitamin D (10.8 ng/ml) and parathyroid hormone levels (11 pg/ml). The level of creatine kinase (CK) which is a muscle-specific enzyme was normal (62 IU/L), and myositis-specific antibody (Jo-1) was negative. Anti-nuclear antibody (ANA), anti-Smith antibody, anti-Ro/SSA antibody, anti-La/SSB antibody and anti-ribonucleoprotein antibody were negative. Mother also had normal serum P, Ca, 1,25-dihydroxy-vitamin D and CK levels and negative autoantibodies including ANA, Jo-1. Of note, both patient and mother were rheumatoid factor (RF) positive. To rule out possibility of nonphosphatemic familial tumoral calcinosis (NFTC) genetic analysis was performed for Sterile Alpha Motif Domain Containing 9 (SAMD9) mutation which is known as key mutation of NFTC, the result was negative.
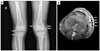
Plain radiographs and magnetic resonance imaging (MRI) revealed multi-nodular extensive soft tissue calcification at the mainly anterior aspect of knee joints of the patient (Fig. 3). There was no evidence of muscle involvement in MRI. Electromyography and muscle biopsy were not performed.
Both patient and her mother were diagnosed with amyopathic JDM based on histopathology and characteristic inflammatory cutaneous manifestations with no clinical evidence of muscle weakness and no serum muscle enzyme abnormalities. Tumoral calcium deposits with "milk of calcium" observed in the patient was diagnosed as dystrophic calcinosis which can be rarely seen in amyopathic JDM.
The patient is being treated with oral acetazolamide (40 mg/kg/d) to manage calcinosis, and the mother is being treated with oral hydroxychloroquine.
DISCUSSION
Amyopathic JDM is an uncommon variant of JDM and is easily under-recognized4. A definition of amyopathic JDM is biopsy confirmed hallmark cutaneous features of DM with onset of under 18 years of age for at least 6 months without clinical or laboratory evidence of muscle disease45. Classical inflammatory cutaneous manifestations of DM include Gottron's papules, periorbital erythema, periungual telangiectasia, and symmetrical violaceous erythema overlying extensor arms and in a shawl-like distribution. No consensus has been made about the minimum condition of skin manifestations required to satisfy the definition of classical or hallmark cutaneous manifestations of DM4. In our case, both the patient and her mother had inflammatory skin manifestation such as Gottron's papule and symmetrical erythematous patch on extensor side of arms since their childhood. And they showed no symptom of muscle weakness and no abnormality of muscle-specific enzyme and myositis-specific antibody. Histopathologic findings of them showed basal layer hydropic changes which can be seen in DM. Autoantibodies including ANA were negative in the patient and mother except for RF. The results of positive RF in both patients strongly indicated an underlying ACTD. In a systematic review of 68 cases of amyopathic JDM, a positive ANA titer was present in about one-half of patients and no mention was made of RF4. However, RF was also observed in patients of JDM as in a previous report6.
Calcinosis is a frequent complication of classic JDM, which occurs in more than one third of patients with JDM1. In contrast, calcinosis in amyopathic JDM is rarely seen. Only 3 cases of 68 (4%) with amyopathic JDM in the systematic review reported calcinosis. Of note, all of these 3 cases evolved into classical DM4. Plain radiographs are generally sufficient for identifying calcinosis, and MRI can help to see the relationship to the surrounding soft tissue7.
The chief complaint of our patient was tumoral deposition of calcium, and she was initially diagnosed at our clinic although her skin manifestations started 5 years ago. Calcinosis can be observed as an initial complaint before diagnosis of JDM8, and diagnosis of JDM can be delayed. Delayed treatment of JDM may be one of the risk factors of calcinosis9, and vice versa. Although delayed treatment of JDM has been mentioned as one of the risk factors which cause calcinosis, early treatment with systemic steroid has not been shown to ensure prevention of calcinosis in a previous prospective study10.
Calcinosis associated with DM is dystrophic calcinosis which is characterized by normal calcium and phosphate levels in serum1 and can be categorized into five subtypes11: (1) small and hard plaques or nodules just below the skin surface; (2) large tumorous deposition which often appear 'popcorn-like' on X-ray examination; (3) deposits in the intermuscular fascia with limitation of movement in the involved muscle group; (4) a severe form which resembles an exoskeleton; and (5) a mixed form of calcinosis.
Tumoral calcium deposition in JDM can be shown in 20% of calcinosis in JDM1213, and when fluid of calcium is collected in pseudobursae, "milk of calcium" can be observed as in our case714. Tumoral calcium deposition should be differentiated from primary tumoral calcinosis (TC), especially normophosphatemic type. TC is a rare disease with the deposition of calcified masses in cutaneous and subcutaneous tissues in peri-articular area. TC is divided into primary TC and secondary TC which is combined with underlying calcifying disease such as chronic renal failure. Serum phosphate levels demarcate the two major subtypes of primary TC, hyperphosphatemic FTC and NFTC15. NFTC is characterized by the absence of metabolic abnormalities and dystrophic calcinosis and NFTC initially manifests with a nonspecific erythematous rash during the first year of life. NFTC is considered to be exceedingly rare, with only six families of Jewish-Yemenite origin reported in literature15. SAMD9 mutation was found in all six families and SAMD9 may play a role in ectopic calcification as an interferon-gamma responsive protein16. However, our patient was negative for SAMD9 mutation, which suggests different pathomechanism in different ethnic groups.
Repetitive trauma, active inflammation, and calcium metabolism abnormality have been suggested as pathogenic factors of calcinosis in JDM17. Dystrophic calcification in JDM can occur anywhere, but large tumorous depositions tend to occur in traumatic locations12. Our patient who has large masses around left knee joint also had left patella fracture three years ago.
Management of calcinosis is challenging, and it should be accompany avoidance of trauma. Medical treatment strategies can be divided into anti-inflammatory drugs such as intravenous immunoglobulin18 and drugs which affect calcium metabolism including diltiazem, aluminum hydroxide suspension, probenecid and colchicine13. Although many treatment options have been suggested to control calcinosis in JDM, no therapy has proven to be effective13. We tried carbonic anhydrase inhibitor acetazolamide which induces phosphaturia by proximal tubular renal acidosis. Acetazolamide may have a valuable synergistic effect in TC19.
Surgical excision of localized calcium deposits may be successful3, but generally excision is not recommended as a treatment of choice because of high risk of poor wound healing and possibility of recurrence and infection20. Furthermore, even biopsy for TC is better avoided for fear of infection. In our case, cellulitis occurred at the site of biopsy on patient's knee (Fig. 3B).
The present case is peculiar in that it is a rare case of TC in amyopathic JDM and that it manifested as an initial presentation of amyopathic JDM. Dermatologists should be aware that calcinosis could be an initial presentation of amyopathic JDM and patients with amyopathic JDM especially combined with calcinosis need to be monitored closely for development of myositis, evolving into classical DM.

 XML Download
XML Download