

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cutaneous and systemic plasmacytosis (CSP) is a rare disorder that occurs mainly in Asians. It is characterized by multiple extensive reddish-brown plaques showing polyclonal plasma cell infiltrates, and various extracutaneous involvements including lymphadenopathy and polyclonal hypergammaglobulinemia. The origin and exact pathogenesis are poorly understood.
This disorder has been grouped with other nonneoplastic lymphoplasmacytic disorders that include systemic plasmacytosis and some variants of Castleman's disease (CD). The distinction between these entities is not well defined because many patients with cutaneous plasmacytosis have variable systemic involvement and overlapping features with others. In this article, we describe the first Korean case showing skin manifestation of typical CSP with renal amyloidosis.
CASE REPORT
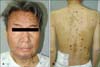
A 54-year-old Korean man was referred to our department for the evaluation of multiple reddish-brown macules and plaques on the face, neck, and trunk (Fig. 1). The pinkish-brown macules were first noticed on the face about 5 years ago, which became hyperpigmented, enlarged, and had spread to the neck and trunk. A few of these macules showed partial spontaneous resolution; however, new lesions appeared constantly. He had renal insufficiency for 5 years and had been managed with peritoneal dialysis three times a week for the last 2 years. However, he had not sought accurate medical advice for his kidney problem and skin lesions.
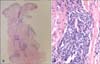
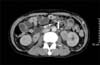
On physical examination, reddish-brown coalescing macules forming reticulated patches were observed over his whole face, and multiple, well demarcated, symmetric dark brown, pea- to bean-sized macules and plaques were found disseminated on his neck, chest, and back. He had an otherwise non specific state except for an anemic appearance and coarse breath sound heard in the chest. Neither hepatomegaly nor splenomegaly was detected, and superficial lymph nodes were not palpable. On laboratory evaluation, complete blood cell count with a differential was notable only for normocytic anemia with a hematocrit of 17.2% (reference, 38%~52%). Moreover, his blood urea nitrogen was 40.1 mg/dl (reference, 0~27.0 mg/dl) and his creatinine was 3.4 mg/dl (reference, 0.4~1.3 mg/dl). Multiple biopsies from the face and back were performed, which revealed superficial and deep perivascular and periadnexal lymphoplasmacytic infiltration in the dermis (Fig. 2A, B). Immunohistochemical staining showed polyclonal reactivity for kappa and lambda immunoglobulin light chains. Serum and urine protein electrophoresis demonstrated polyclonal elevation of gamma globulins. A computed tomography scan of the abdomen revealed diffuse lymphadenopathy along the para-aortic, aortocaval, both renal hilar, obturator, and inguinal lymph nodes (Fig. 3). A bone marrow biopsy specimen and aspiration showed normocellular marrow with scattered polyclonal plasma cells representing about 7% of marrow cellularity. Deposition of homogeneous eosinophilic materials was seen at the glomerulus on renal biopsy (Fig. 4A). A Congo red-stained section revealed yellow-green birefringence under polarized light (Fig. 4B). Congo red stain is the most popular method for detecting amyloid. On electron microscopy, the bulk of the amyloid was found in the mesangium.
He received a regimen of 10 mg/day prednisolone and 1,000 mg/day mycophenolate mofetil with hemodialysis to control the renal insufficiency, 3 times a week. After 5 months of treatment, the reddish-brown macules on the face, neck, and trunk showed moderate regression leaving a slight pigmentation. Unfortunately, after 1 year, he suddenly died at home. Post-mortem examination was not carried out.
DISCUSSION
CSP was first described by Yashiro1 in 1976 and further refined in 1980 by Kitamura et al.2 by using the term "cutaneous plasmacytosis." It has been proposed that patients with cutaneous plasmacytosis should be evaluated for systemic involvement, even in the absence of symptoms3, and the condition has been subsequently named "cutaneous and the systemic plasmacytosis" to emphasize the significant frequency of extracutaneous involvement4. It is described primarily in middle-aged Asian patients and in particular in Japanese patients. Previous reports about CSP have been restricted to patients of Asian ancestry and, to our knowledge, five cases have been reported in the Korean literature.
CSP is characterized by cutaneous polyclonal plasma cell infiltrates associated with various extracutaneous involvements including lymphadenopathy and polyclonal hypergammaglobulinemia. In most cases, the clinical presentation is very characteristic, with multiple reddish-brown infiltrated macules, plaques, and flat tumors mainly on the trunk. The disease is sometimes accompanied by anemia and fever5.
The origin and exact pathogenesis are poorly understood; however, current evidence suggests that a deregulated production of interleukin (IL)-6 plays an important role in the pathogenesis. IL-6 is a cytokine that induce B-cell proliferation and differentiation to plasma cells, immunoglobulin secretion, and angiogenesis67.
Histologically, CSP shows a moderately dense superficial and deep perivascular infiltrate of predominantly mature plasma cells without atypia. Variable numbers of mature lymphocytes may be present8. Lymphoid follicles with reactive germinal centers were seen in some cases5. Plasma cells usually revealed a polyclonality expression of both kappa and gamma immunoglobulin light chains9. Plasmacytosis should be differentiated from other conditions resulting in the proliferation of plasma cells, including chronic infection, collagen vascular disease, cutaneous marginal B-cell lymphoma, and B-cell pseudolymphoma. Although the plasma cells are usually polyclonal in CSP, some cases may indeed harbor monoclonal population of plasma cells5. The relation between CSP and CD is controversial. Except for the skin lesions, CSP and CD share similar clinical and histological features. The histological features of CSP showed infiltration of mature plasma cells, similar to multicentric plasmacytic CD (MPCD). Overproduction of IL-6 in the serum has been reported and considered an important factor in the pathogenesis of MPCD. IL-6 is also highly expressed in the serum of some patients with CSP. On the basis of the similar clinical and histological features, it has been suggested that CSP and cutaneous MPCD may be considered a disease continuum rather than distinct entities, which needs further studies to clarify10.
The most common extracutaneous involvement is lymphadenopathy, which occurs in approximately 58% of patients. Lung involvement presents as lymphoid interstitial pneumonia. Liver, spleen, and kidney involvement occur less frequently4. In the Korean literature, there has been no case of CSP with involvement of such a variety of organs. Renal involvement in CSP is highly heterogeneous and may be coincidental in many cases. All cases of renal amyloidosis have been known as secondary amyloidosis11. Our patient had no history of recurrent infections, chronic inflammatory diseases, autoimmune disease, or neoplasm. We could not find other causative factors. In our patient, CSP was associated with renal amyloidosis. CD has been associated with renal amyloidosis in very few patients; however, we could not find any evidence in the literature on an association between CSP and renal amyloidosis10. Amyloidosis may progress rapidly in cases with high serum amino acids and C-reactive protein. IL-6 may also play a role in amyloid deposition. The overproduction of IL-6 in the serum considered an important factor in the pathogenesis of CSP, similarly to MPCD. It is possible to postulate that a relation exists between IL-6 and amyloid production and deposition in CSP.
Most cases of plasmacytosis have followed a chronic and benign clinical course without spontaneous remission9. In fact, although there are reports of patients with plasmacytosis developing malignant lymphoma such as T-cell lymphoma and non-Hodgkin lymphoma, the relation between plasmacytosis and lymphoma is not clear. However, there is a low rate of malignant transformation of CSP in Asian studies. A single case of T-cell lymphoma arising in a patient with CSP has been reported; however, whether this was a coincidental finding is unclear9.
Various treatments have been tried, with no one modality affording consistent clinical remission. The treatment approaches including topical and systemic corticosteroids, antibiotics, and systemic chemotherapy has shown to be of some benefit; however, the results were poor. Topical 0.1% tacrolimus ointment has been shown to help reduce the thickness and pigmentation of skin lesions8. Photodynamic therapy decreased the thickness of lesions, whereas long-pulse ruby laser helped lighten the pigmentation8. A recent report described treatment success with a combination of prednisolone and cyclophosphamide. Radiotherapy, phototherapy, and intralesional corticosteroids have been shown to improve cutaneous lesions4. However, the poor treatment response of this disorder clearly demonstrates the needs for a novel therapeutic method.
Although most patients with CSP experience a chronic benign course without spontaneous regression, a few cases had an aggressive clinical course with a fatal outcome. Therefore, special attention should be given to monitoring for pulmonary and renal involvement.

 XML Download
XML Download