

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Over the past 25 years, enormous advances have been made in understanding the clinical and molecular basis of epidermolysis bullosa (EB); new phenotypes have been described, molecular insights have been gained, and translational research has launched innovative clinical trials of new cell- and gene-based therapies1. Summarizing recent data, a schematic of the protein/structural pathology currently implicated in the pathophysiology of EB is shown in Fig. 1. Concerning disease taxonomy, several attempts have been made to classify and reclassify EB, with helpful guidelines published by an international consensus group2. Although elements of the current classification of EB are still based on the level of blister formation at or within the dermal-epidermal junction or epidermis, these latest guidelines introduced an "onion skin" classification, providing a workable diagnostic algorithm based on variable access to general or specialized skin and molecular laboratory tests2. Most people with EB can now be classified into one of the existing disease categories, although, over the past few years several new clinicopathologic entities have been added to the collection of EB disorders. These newly reported conditions are often very rare and affect just a few individuals or families. Nevertheless, identification and recognition of these distinct clinicopathologic entities is important in improving clinical skills and gaining better insight into the pathophysiology of skin blistering and the macromolecular intricacies of adhesion and signaling that maintain healthy skin3. This review focuses on three relatively recent discoveries in EB. All three disorders are autosomal recessive, and comprise two forms of EB simplex and one of junctional EB with extracutaneous abnormalities, which result from biallelic mutations in DST-e, EXPH5, or ITGA3, respectively. This review describes the discovery of these conditions, outlines the clinical manifestations, summarizes the current known mutations, and places the discoveries in the context of skin pathology and the pathophysiology of blistering.
DST-e: autosomal recessive epidermolysis bullosa simplex
Pathogenic mutations in DST-e were first reported in 20104. Their discovery followed identification of a parent-related Kuwaiti family with a relatively minor form of acral skin blistering. The clues to diagnosis came from transmission electron microscopy: the patient's skin was completely lacking in hemidesmosomal inner plaques, raising the possibility that the inherent gene pathology lay in a protein normally sited within this part of the hemidesmosomal complex. A candidate gene approach with immunofluorescence microscopy and antibodies to various hemidesmosomal inner plaque components demonstrated a complete absence of immunostaining for the 230 kDa bullous pemphigoid antigen (BP230). Sanger sequencing of DST-e, which encodes the BP230 protein, also known as epidermal dystonin, revealed a homozygous nonsense mutation, thus establishing a new form of autosomal recessive EB simplex with loss-of-function mutations in DST-e.
DST-e: clinical features
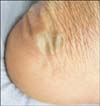
To date, six individuals from six different families with autosomal recessive EB simplex due to DST-e pathology have been reported; five individuals were from Kuwait, the other patient was from Iran. All were from parent-related families. In most cases, blistering began at or shortly after birth. The blistering differed from more classic localized forms of EB simplex associated with mutations in keratin 5 (KRT5) and keratin 14 (KRT14), on the basis that some of the blisters were not directly related to pressure points on the soles of the feet or toes and the blisters were somewhat larger (up to several centimeters in diameter). The sides or dorsae of the feet were often affected (Fig. 2). There were also a few other scattered generalized blisters, but overall the clinical features were of mild severity. No ocular, genitourinary, or other mucosal abnormalities have been observed. Blistering has tended to heal without scarring, but in the affected Middle-Eastern patients, post-inflammatory hyperpigmentation was common. Although the condition is autosomal recessive, a semi-dominant transmission mode is also plausible, given that some heterozygous carriers (e.g., parents of affected cases) recall some blistering in childhood that usually improves and then stops completely by the teenage years56. Notably, the father of one the Kuwaiti patients reported some blistering during childhood that resolved in adulthood, and both heterozygous children of an affected Iranian woman had minor skin blistering.
DST-e: molecular pathology
Thus far, two pathogenic homozygous nonsense mutations in DST-e have been identified45. The first case reported was in a parent-related Kuwaiti family4, with Sanger sequencing of genomic DNA revealing homozygosity for p.Gln1124*. The DST gene encodes multiple transcripts with different tissue expression patterns, but the mutation identified in this case is specific to the epidermis, which is consistent with the lack of extracutaneous clinical findings. The second mutation identified was p.Arg1249*. It was reported in an Iranian woman born to consanguineous parents5. This mutation is also located within the coiled-coil domain of the epidermis-specific isoform of dystonin. Subsequently, four other individuals from Kuwait6, who were not known to be related to one another, presented with similar blistering and were all shown to be homozygous for p.Gln1124*, the mutation that was originally identified in a Kuwaiti patient. Haplotype analysis in the five pedigrees revealed a shared ~60 kb block of genomic DNA across the site of the mutation, consistent with a founder effect6.
DST-e: skin pathology
Strikingly, loss-of-function mutations in DST-e give rise to major ultrastructural abnormalities with a complete absence of the hemidesmosomal inner plaque (Fig. 3), a key attachment point for keratin intermediate filaments and the location for other major plaque proteins, such as plectin45. Therefore, it is perhaps somewhat surprising that clinical features associated with complete loss of DST-e present with such relatively minor blistering, a finding that contrasts with the blistering found among many patients with the acquired autoimmune condition bullous pemphigoid who have autoantibodies to BP230 (although many of the latter also have pathogenic antibodies that target the 180 kDa bullous pemphigoid antigen). Studies of keratinocytes from patients with DST-e mutations have identified loss of adhesion and increased cell spreading and migration7. Mutant cells also display reduced β4 integrin at the cell surface and have increased total protein levels of keratin 14 and β1 integrin7. The upregulation of keratin 14 expression at the protein level might offer partial compensation, which may help to explain the relatively mild blistering phenotype associated with loss of DST-e expression. Collectively, these cell biology studies identify a previously unexpected role for DST-e in regulating keratinocyte adhesion and in controlling functional switching between integrin subtypes in keratinocytes.
DST-e: clinicopathologic comments
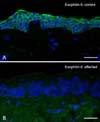
The limited degree of predominately acral skin blistering may make it clinically difficult to distinguish autosomal recessive EB simplex with DST-e pathology from other forms of EB simplex, although, the presence of larger acral blisters in non-pressure prone sites may be indicative. However, the lack of hemidesmosomal inner plaques on transmission electron microscopy gives the clearest clue to the underlying diagnosis. Alternatively, immunostaining of the patient's skin with an antibody to DST-e (BP230) is also likely to show a marked reduction in or complete absence of immunostaining, thereby highlighting the candidate gene for mutations and the likely diagnosis (Fig. 4).
EXPH5: autosomal recessive epidermolysis bullosa simplex
The identification of EXPH5 as a potential gene for EB was made by next-generation sequencing8. Three affected children from a parent-related Iraqi family presented with superficial generalized blistering with histological evidence of intraepidermal skin fragility and keratin tonofilament disruption, predominately in basal keratinocytes. Sanger sequencing of known genes for EB simplex, KRT5, KRT14, and PLEC (plectin), failed to identify pathogenic mutations; therefore, whole exome sequencing was performed, with a focus on identifying novel homozygous, potentially damaging variants. A homozygous frame-shift mutation in EXPH5 was identified. EXPH5 encodes exophilin-5, also known as Slac2-b, a Rab27 GTPase with a role in intracellular vesicle transport to the cell membrane9.
EXPH5: clinical features
So far, seven cases of autosomal recessive EB simplex due to EXPH5 pathology have been reported in four different families from several countries: Iraq, Germany, the United Kingdom, and Pakistan. The initial parent-related Iraqi family contained eight siblings, of whom three had similar skin fragility8. Starting in early life, affected individuals had trauma-induced scale crusts and intermittent blistering (Fig. 5). Intact blisters were often rare, but the siblings typically presented with trauma-induced grazes and scale crusts that were sometimes slow to heal. Affected sites recovered with very slight scarring and post-inflammatory hyperpigmentation. There were no extracutaneous abnormalities. In all patients, blistering tended to decrease with age. The second individual, from a non-parent-related European family, presented with more generalized blistering and large erosions that subsequently became milder, with later lesions predominantly consisting of acral vesicles10. In the third case, also from a European family, blistering only became evident after several months when the child started to walk11. He experienced widespread small erosions and bleeding, the major areas affected being the lower back, knees, and ankles. There were no intact blisters and he was able to tolerate sticking plasters without harm. In the fourth family, from Pakistan, two siblings had scattered vesicles and blisters from birth, with individual lesions healing within one to two weeks to leave post-inflammatory hypopigmentation12. With increasing age, the blistering tendency gradually improved and almost remitted.
EXPH5: molecular pathology
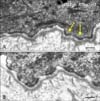
Six different frameshift or nonsense mutations in EXPH5 have been reported thus far. The first mutation, identified in the Iraqi family, was a homozygous frameshift mutation, c.5786delC (p.Pro1929Leufs*8). Then, two European cases were shown to be compound heterozygotes for biallelic loss-of-function mutations: the mutations in the German case were c.1395delC (p.Phe466Leufs*44) and c.2897delC (p.Pro966Leufs*11), whereas in the British case the mutations comprised c.1947dupC (p.Pro649Profs*11) and c.2249C>A (p.Ser750*). The most recently reported case, from Pakistan, involved homozygosity for c.3650T> A (p.Leu1217*). All pathogenic mutations were located within exon 6 and complete loss of exophilin-5 immunostaining was observed in all three of the cases for which immunofluorescence microscopy was performed. In each case, the route to molecular diagnosis followed different strategies: The first case involved whole exome sequencing. The second case was extracted from a panel of 70 undiagnosed cases of EB simplex (i.e. previously negative for mutations in KRT5, KRT14, and PLEC). In the third case, the clue to diagnosis came from transmission electron microscopy, which revealed large perinuclear collections of vesicles in basal keratinocytes (Fig. 6). In contrast, the fourth case was diagnosed clinically on the basis of a spontaneously improving, autosomal recessive blistering disorder during childhood.
EXPH5: skin pathology
The nature of the pathology in this subtype of EB simplex appears to be somewhat different from other EB simplex entities in that exophilin-5 is (a) not a structural protein and (b) not an integral component of intermediate filaments, hemidesmosomes, or desmosomes13. Indeed, the precise role of exophilin-5 in keratinocytes is unknown: it is a Rab27B GTPase, with a proposed contribution to the regulation of cell membrane traffic, including vesicle formation and vesicle movement along actin and tubulin networks. Knockdown experiments in HaCaT cells have shown a key role for transport of some vesicles to the cell membrane, leading to speculation about keratinocyte integrity being compromised as a result of defective exosome trafficking and release of certain proteins, RNAs, and microRNAs from the cell14. Knockdown of EXPH5 also leads to reduced cell adhesion to the basement membrane, providing an explanation for the observed skin fragility8. In terms of the cell pathology, there is marked disruption of the keratin filament network. Indeed, some of the keratin filament aggregation shows a basket-weave pattern and ball-like aggregates that are reminiscent of generalized severe EB simplex associated with dominant-negative mutations in KRT5 or KRT14. Exophilin-5 only colocalizes with Rab27B in certain compartments of the cell and appears to have a role in controling β4 integrin function in keratinocytes8. It is not presently known how this particular GTPase affects the microtubule and keratin filament interaction. Future identification of key proteins involved in this connectivity will most likely provide fascinating new insights into both the pathophysiology of skin blistering and skin homeostasis.
EXPH5: clinicopathologic comments
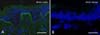
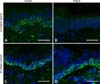
Clues to the diagnosis of this subtype of autosomal recessive EB simplex can come from both clinical and skin biopsy evaluation. First, the finding of relatively mild blistering in someone with a pattern of inheritance consistent with autosomal recessive transmission provides a good clue. Indeed, there is also a natural history for the skin blistering to improve or remit during childhood. Ultrastructurally, the skin may show abnormal collections of cytoplasmic vesicles and, where available, immunostaining with an antibody to exophilin-5 may show a complete absence of skin staining (Fig. 7). For next-generation sequencing approaches, biallelic loss-of-function mutations in EXPH5 will help uncover this diagnosis.
ITGA3: junctional epidermolysis bullosa
A candidate gene approach was also used to discover the first pathogenic mutations in ITGA3, although, the skin component of the underlying disorder was minor in comparison to the major inflammation affecting the lungs and kidneys15. The initial case had minor blistering, and transmission electron microscopy showed cleavage within the lamina lucida, some lamina densa reduplication, and the complete absence of immunostaining for the α3 integrin subunit in the basement membrane of the skin. Sanger sequencing subsequently confirmed loss-of-function mutations in ITGA3. Following diagnosis of the initial case, two more cases, presenting with nephrotic syndrome, pulmonary inflammation, and minor skin blistering, were also Sanger sequenced and further mutations were identified15. Subsequently, two further cases have been reported, although neither had skin blistering as part of the phenotype1617.
ITGA3: clinical features
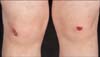
To date, only five cases of EB related to mutations in ITGA3 have been described; all presented as very sick neonates and the skin was not the major health concern151617. The first neonate presented within a few hours of birth with respiratory difficulty and required assisted ventilation. Within two weeks, this neonate developed renal failure with massive proteinuria. However, skin fragility did not manifest until after three months, with small blisters and erosions occurring after minor trauma. The child died before eight months of age because of pulmonary infection. A second neonate presented with respiratory distress at 48 hours of life, developed nephrotic syndrome at six weeks of life, and died from multi-organ failure aged two months; skin fragility was not highlighted in this case. The third patient developed fever and respiratory distress at two months of age, proteinuria at five months, and blisters on the shins (initially manifesting as erythematous macules) from the age of four months. The child died aged 17 months from multi-organ failure. For the causative gene abnormality in these cases, it was evident from the symptoms that the candidate gene was expressed in the kidneys, lungs, and skin. However, the fourth case presented with respiratory distress at birth and nephrotic syndrome but no blisters16. There was interstitial lung disease and unilateral renal hypoplasia and hydronephrosis; the child died from respiratory failure aged 7 months. The fifth case presented with respiratory distress at 2 weeks of age requiring ventilator support17. There was interstitial lung disease, chest infections and renal anomalies (crossed fused renal ectopia) and nephrotic syndrome; dermatologic changes were limited to sparse hair and toe nail dystrophy which were evident from five months. The child died before the age of 7 months from pulmonary infection.
ITGA3: molecular pathology
The first three described cases of EB related to mutations in ITGA3 were published together in a single report15. All patients were from parent-related families and were identified has having homozygous mutations. The first case, from Southern Italy, had a homozygous frameshift mutation, c.1173_1174del (p.Pro392Valfs*2). The second case, from Gaza, had a homozygous acceptor splice site mutation, c.1538-1G>A. The third case, from Pakistan, had a homozygous missense mutation, c.1883G>C (p.Arg628Pro) that occurred within the extracellular domain of α3 integrin, which may also compromise exon splicing. The mutation in the fourth case (from The Netherlands) was also a homozygous missense mutation, c.1045G>T (p.Ala-349Ser)16, as was the mutation in the fifth case, c.1387C>T (p.Arg463Trp)17. Clues to the molecular pathology of the first case came from skin histology and immunohistochemistry, with evident disruption of the basement membrane and a complete absence of immunostaining for α3 integrin15. The other two cases in the initial report were diagnosed by screening DNA for mutations in ITGA3 from a DNA archive of eight unrelated patients with congenital nephrotic syndrome15. Case four was diagnosed following exclusion of other candidate genes, homozygosity mapping and direct Sanger sequencing of ITGA316, whereas in case five, skin immunohistochemistry was used to identify α3 integrin as the candidate gene despite the lack of skin fragility clinically17.
ITGA3: skin pathology
The alpha-3 integrin subunit normally dimerizes with the β1 integrin subunit within focal contacts at the dermal-epidermal junction18. In the first patient with loss-of-function mutations in ITGA3, transmission electron microscopy showed some structural similarities with those previously reported in α3 integrin knockout mice19. Notably, the patient's skin had a thin, discontinuous lamina densa. Although the lamina densa was typically located in the blister floor, it was occasionally adhered to cell fragments, indicating multiple levels of cleavage in and around the basement membrane15. Although the skin pathology showed some histological overlap with Kindler syndrome, the candidate gene was identified by the complete lack of α3 integrin immunoreactivity in the patient's skin. Initial skin biopsy findings from patient three in the initial report that are presented for the first time in this review, revealed cleavage mostly within the lamina lucida and focal reduplication of the basement membrane, but normal hemidesmosome-anchoring fibril complexes (Fig. 8). In this case, immunostaining for the α3 integrin subunit was completely absent, in contrast to a normal staining intensity and pattern for the β1 integrin subunit (Fig. 9), consistent with a capacity for β1 integrin to dimerize with other integrin subunits20. Functional studies in three cases have demonstrated the deleterious effects of the mutations (p.Ala349Ser, c.1538-1G>A, and p.Arg463Trp) on α3 integrin structure, processing and dimerization161721.
ITGA3: clinicopathologic comments
Clues to diagnosing this rare form of EB relate to the major clinical features of lung and kidney inflammation22. In the presence of severe respiratory distress, lung infiltration and infection, as well as nephrotic syndrome and renal failure, and the finding of minor skin blistering that starts between two to four months of life, then skin immunostaining for α3 integrin or Sanger sequencing of ITGA3 is recommended for candidate gene screening. The constellation of clinical features in this genodermatosis reflects the predominant tissue expression of α3 integrin in the kidneys, lungs, and skin. Nevertheless, it is clear that skin blistering is either mild or absent in this disorder and thus whether the phenotypic consequences of ITGA3 mutations should remain as part of the designated spectrum of EB, defined as a group of inherited blistering skin diseases, or not, is debatable.
CONCLUSIONS
This review has described the salient clinical and molecular features associated with three recently described forms of EB. Although these disorders are rare, understanding their nature allows their recognition as distinct clinicopathologic entities and their clinical diagnosis in similar cases. Moreover, all cases are associated with loss-of-function mutations, giving some indication as to the human consequences of complete loss of expression of these specific proteins. Thus, the clinical, pathology, and molecular data provide an improved understanding of the mechanisms underlying skin integrity, whilst highlighting some of the compensatory processes that may occur. In future, it is most likely that a small number of other novel forms of EB will be identified that further add to what is known about the pathophysiology of skin blistering.



 XML Download
XML Download