

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor:
Livedoid vasculitis is a rare disorder clinically characterized by purpuric macules and papules on the lower legs and feet1. These lesions are caused by infarction of small vessels that affects young and middle-aged women. The bizarrely shaped ulcers may be painful, heal slowly, result in scarring, or cause atrophie blanche2. Livedoid vasculitis usually occurs in isolation; however, several reports have shown an association with systemic lupus erythematosus, other autoimmune diseases, and malignant disease3. However, an association with antiphospholipid antibodies has been rarely described.
Antiphospholipid syndrome is an acquired hypercoagulable state, shows recurrent thrombosis, and can spontaneously resolve. It is correlated with systemic lupus erythematosus in about half of the patients. There are only a few reports about patients with livedoid vasculitis accompanying primary antiphospholipid syndrome without systemic lupus erythematosus. Primary antiphospholipid syndrome could be diagnosed by using the criteria of Miyakis et al.4 (small vessel thrombosis, spontaneous abortion/anticardiolipin antibody subclass immunoglobulin (Ig) G or IgM, anti-β2 glycoprotein IgG or IgM, and lupus anticoagulant).
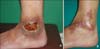
A 33-year-old female patient with painful tender erythematous ulcers on both lower legs visited our hospital in December 2012. The ulcers had been covered with exudate and crust for 4 months. She had been treated with a nonsteroidal anti-inflammatory drug for several months before visiting our institution. Irregular ulceration and purpura were observed on admission (Fig. 1A). Histological examination showed evidence of dilated capillaries in the upper dermis and thickened vessel walls containing fibrinoid materials (Fig. 2). She was initially treated with systemic methylprednisolone, but she failed to respond. Screening for vasculitis gave normal results, including lupus relevant antibodies (lupus anticoagulant, antinuclear antibody, and anti-dsDNA antibody), except for the increase in anti-cardiolipin antibody subclass IgG at 17.2 U/ml (reference, <9 U/ml) and a markedly increased anti-cardiolipin antibody subclass IgA at 20.8 U/ml (reference, <10 U/ml). Additionally, she had increased anti-phospholipid antibody subclass IgG at 18.4 U/ml. Therefore, the diagnosis was concluded to be livedoid vasculitis and primary antiphospholipid syndrome, and she was started on combination therapy with 100 mg aspirin and low-dose warfarin (2 mg) daily with a good clinical response (Fig. 1B). She remains in remission presently.
The treatment for livedoid vasculitis with antiphospholipid syndrome is not clearly defined. Corticosteroids have been used successfully5; however, they do not provide long-term benefits, and our patient showed no response. Antithrombotic agents, including tissue plasminogen activator, prostacyclin, antiplatelet therapy, and low-dose warfarin, can be used successfully123. The combination of low-dose warfarin and antiplatelet therapy was used in our patient to prevent further thrombosis.
The patient demonstrated histologically thickened vessel walls containing fibrinoid material and clinically irregular ulcerations and purpura. Therefore, her condition was diagnosed as livedoid vasculitis with primary antiphospholipid syndrome and she was treated with low-dose warfarin, an antiplatelet agent.
Livedoid vasculitis presents the clinical symptoms of other diseases that cause occlusive vasculopathy; thus, clinicians should find the underlying conditions and provide treatment if antiphospholipid antibodies are present. Aspirin and low-dose warfarin combination therapy is a valuable therapeutic option for livedoid vasculitis with primary antiphospholipid syndrome.

 XML Download
XML Download