

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Hemophagocytic lymphohistiocytosis (HLH) is a rare and fatal disorder. The initial clinical presentations include fever, hepatosplenomegaly, and pancytopenia. Additional symptoms include lymphadenopathy, pulmonary infiltration, neurologic abnormalities, and dermatologic manifestations1234. The cutaneous manifestations are variable and nonspecific13. Although purpuric, maculopapular, erythrodermic, or morbilliform eruptions have been reported as the cutaneous manifestations of HLH, there was no reported case showing annular skin lesions with bullae associated with HLH123. Herein, we report an interesting case of HLH with an initial clinical manifestation of annular skin lesions that subsequently changed to bulla formation.
CASE REPORT
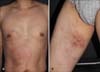
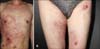
A 42-year-old man presented with painful multiple annular or arcuate, erythematous to purpuric and pigmented patches on the trunk and right thigh for 3 months (Fig. 1). He had a history of hepatitis B and denied taking any medication. The clinical impression was erythema annulare centrifugum (EAC) or urticarial vasculitis. After obtaining informed consent from the patient, skin biopsies were performed on the chest and right thigh for the diagnosis. Histopathologic findings showed a few dyskeratotic cells and basal vacuolar changes in the epidermis. Dermal edema, mild vascular ectasia, and red blood cell extravasation were observed in the upper dermis. Dense perivascular and periadnexal inflammatory cell infiltration and diffuse dermal inflammatory cell infiltration were present in the mid and deep dermis (Fig. 2). These did not fit the typical findings of EAC. With the suspicion of EAC, topical steroids (0.05% desonide lotion and 0.05% desoximetasone gel) and 0.1% tacrolimus ointment were applied for 2 months. However, 2 months later, the patient presented again to our clinic with painful superimposed multiple denuded lesions on the annular patches, which had increased in number. The lesions not only had an erythematous annular ring-shape pattern but also had a violaceous or brownish hue (Fig. 3). Another skin biopsy specimen taken from the bulla revealed epithelial denudation due to nonspecific epidermal necrosis and a perivascular lymphocytic infiltration in the dermis (Fig. 4). Direct immunofluorescence was performed on the perilesional skin to exclude the possibility of autoimmune bullous diseases; however, the result was negative. Cyclosporine (200 mg/day) was started; however, the patient complained of high fever (up to 39℃) 4 days later. The laboratory test showed an elevated aspartate aminotransferase level of 760 UI/L (14~40 UI/L) and alanine aminotransferase level of 488 UI/L (9~45 UI/L). The neutrophil count was 1.84×109/L (4~10×109/L), hemoglobin 8.4 g/dl (13~18 g/dl), platelet count 21×103/ml (150~450×103/ml), triglyceride 364 mg/dl (40~200 mg/dl), fibrinogen 40 mg/dl (160~350 mg/dl), and ferritin 39,980 ng/ml (300~400 ng/ml). The test for anti-nuclear antibody was negative. The reverse transcriptase-polymerase chain reaction (RT-PCR) result for Epstein-Barr virus (EBV) was 1,995,710 copies/ml, indicating an EBV infection. Physical examination also showed not only worsening of the skin lesions but also sudden weight loss (10 kg/3 months) and splenomegaly. The patient was placed on entecavir treatment, and cyclosporine was stopped as it might cause hepatitis B to flare up. At that time, necrolytic migratory erythema (NME) and paraneoplastic pemphigus were suspected because of the annular bullous erythema and systemic symptoms such as weight loss. However, there was no evidence of glucanoma on the abdominal computed tomography (CT). Serum immunoblotting for evoplakin/periplakin was performed to exclude the possibility of paraneoplastic pemphigus; however, the result was negative. A bone marrow biopsy was then performed for the evaluation of pancytopenia. The results showed histiocytic hyperplasia and active hemophagocytosis. On the basis of these findings, EBV-associated HLH was diagnosed. The patient underwent chemotherapy with etoposide and dexamethasone. Six cycles of the chemotherapy were administered. However, the patient did not respond to chemotherapy and he died of septic shock 6 months later.
DISCUSSION
HLH represents a severe and life-threatening hyperinflammatory state. It is not a single disease but a group of conditions leading to the same hyperinflammatory state. It can be classified, according to the underlying etiology, into either primary or familial HLH, and into a secondary disease or infection, malignancy, or rheumatologic disorder associated with HLH. Primary HLH is mainly due to defects in exocytosis-mediated cytotoxicity. The mechanisms underlying secondary HLH, such as EBV-associated HLH and macrophage-activation syndrome, remain unclear23.
The clinical manifestations of primary and secondary HLH overlap. The most common presentations are prolonged fever and hepatosplenomegaly2. Additional symptoms include lymphadenopathy, pulmonary infiltration, neurologic abnormalities, and dermatologic manifestations. It is difficult to diagnose HLH because of its variable clinical presentations. A high degree of clinical suspicion is important in the diagnosis of HLH2. Clinicians should perform laboratory examinations including complete blood count, liver function test, serum triglyceride and ferritin tests, and bone marrow biopsy when considering the diagnosis of HLH in patients showing the above-mentioned symptoms and signs. The Histiocyte Society presented the diagnostic guidelines for HLH in 1991, which were updated in 2004.
Six percent to 67% of HLH patients show skin eruptions1. These cutaneous manifestations are classified into three types: (i) specific to the underlying malignancy (e.g., cutaneous lymphoma or systemic disease); (ii) reflecting the biological consequences of HLH (e.g., thrombopenic purpura or conjunctival jaundice); and (iii) a generalized, transient, nonpruriginous maculopapular rash3. Not only maculopapular rashes but also extensive purpuric macules, generalized erythroderma, and edema have been reported123. Briefly, the cutaneous manifestations are variable and the histopathologic findings are nonspecific. Our case did not fit into any of the three categories mentioned above. To our knowledge, there has been no previous case report of HLH in a patient presenting with annular skin lesions and epidermal necrosis as a dermatologic manifestation.
When a patient presents with annular patches accompanied by bulla and systemic symptoms, paraneoplastic diseases such as paraneoplastic pemphigus and NME, which is a manifestation of the glucagonoma syndrome, should be considered. Histopathologically, necrosis is observed in NME, with separation of the upper layers of the epidermis and neutrophilic infiltration in the necrotic layer5. Abdominal CT findings in conjunction with an increase in fasting plasma glucagon levels to >1,000 ng/L confirm the diagnosis. Paraneoplastic pemphigus is an autoimmune vesiculobullous and erosive mucocutaneous disease associated with underlying malignancies. Its histologic features are variable. The presence of antibodies to envoplakin and/or periplakin is believed to be highly specific for paraneoplastic pemphigus, and the linker domain of plakins may be of particular significance5. In our patient, there was no evidence of glucanoma syndrome or paraneoplastic pemphigus. Additionally, chemotherapy with etoposide and dexamethasone resulted in a marked improvement of skin lesions after the diagnosis of HLH.
Stevens-Johnson syndrome/toxic epidermal necrolysis complicated with HLH may also be considered; however, our patient's skin lesions showed slow progression during 2 months, which is different from the clinical features of this disease. Although EBV-related persistent erythema multiforme has also been reported, it may be ruled out in our case as there is seldom an infiltration of inflammatory cells observed in the pathology of this diagnosis6.
HLH itself can be fatal, and extensive epidermal necrosis accompanied by complications such as fluid and electrolyte imbalance, renal failure, and septicemia due to secondary infection can increase the fatality of the disease7. Thus, effective treatment should be initiated promptly when indicated by clinical suspicion. The therapy involves immunosuppressive drugs and/or chemotherapy. Allogeneic stem cell transplantation is required for the cure of all primary HLH and some cases of very severe secondary HLH24. Our case shows how difficult it is to diagnose HLH if an annular erythema is the only initial sign of the disease. Therefore, a high degree of clinical suspicion is important in the early diagnosis and prompt treatment of HLH.


 XML Download
XML Download