

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Keloids are benign hyperproliferative growths of connective tissue and are composed of large collagen bundles that extend beyond the boundary of the original wound. They are characterized by the proliferation of dermal fibroblasts, overproduction of extracellular matrix components, and increased infiltration of inflammatory cells including lymphocytes, mast cells, and macrophages. Deposition of a dense collagenous meshwork with a rich vascular supply and increased intrusion of inflammatory cells are probably related to the proliferative and inflammatory characteristics of keloids. However, the precise pathogenic mechanisms that cause keloids remain unknown.
Previous studies have investigated the effects of various growth factors and the disruption of apoptotic mechanisms in the pathogenesis of keloids1234. A number of abnormalities in various cellular functions, including proliferation, apoptosis, or expression of growth factors and extracellular proteins, were found in fibroblasts derived from keloid tissues56. Recently, a significant increase in the generation of reactive oxygen species (ROS) was found in keloid fibroblasts, which could relate to the inflammatory and oxidative stress features of the disease7.
Nuclear factor erythroid 2-related factor 2 (Nrf2) is one of the most important cellular defense proteins to combat oxidative stress8-10. A protective role for the Nrf2 pathway has been established in many human disorders, including cancer, inflammatory disease, and neurodegenerative disease11. Moreover, it has been reported that increased free radicals and impaired antioxidant scavenging are involved in the pathogenesis of keloids11.
As Nrf2 is a main defense mechanism against oxidative stress, a decrease in expression of Nrf2 may contribute to increased ROS production in keloid tissue. To the best of our knowledge, there have not been any studies reporting the expression of antioxidant proteins in keloids. Here, we compare Nrf2 expression in normal skin and keloid tissues using western blot and immunohistochemical analyses, and examine its roles in human fibroblast cells transfected with Nrf2-specific small interfering RNA (siRNA).
MATERIALS AND METHODS
Tissue sample collection and preparation
Keloid specimens were obtained from patients who had undergone surgery in the Department of Plastic and Reconstructive Surgery. The institutional review board of Soonchunhyang University Seoul Hospital reviewed and approved this research protocol involving the use of tissue samples.
Eight keloid tissues were obtained from eight female patients and were diagnosed by pathology. Normal skin tissues were collected from the backs of 8 women who had breast reconstruction with a latissimus dorsi flap (Table 1). For immunohistochemical analysis, archival formalin-fixed, paraffin-embedded tissues were used.
We used 2~3 specimens from each of the eight normal skin samples and eight keloid samples. A portion of each specimen was frozen in liquid nitrogen immediately after resection for western blotting analysis, and stored at -80℃. The remaining samples were fixed in formalin, embedded in paraffin wax, and then stored until needed for immunohistochemical studies.
OxyBlot (measurement of protein carbonyl by immunoblotting of the 2,4-dinitrophenylhydrazine-protein)
The OxyBlot Protein Oxidation Detection kit (Chemicon International, Temecula, CA, USA) was used to detect carbonyl groups introduced into proteins by oxidative modification. 2,4-Dinitrophenylhydrazine (DNP) derivatization was carried out for 15 min following the manufacturer's instructions on 10 µg of protein obtained from the tissue. The DNP-derivatized protein samples were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to polyvinylidene difluoride membranes, stained with Ponceau red, and then probed with an anti-DNP antibody. Blots were developed using a chemiluminescence detection system.
Western blot analysis
Tissue samples were homogenized in whole cell extract buffer [25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (pH 7.7), 0.3 M NaCl, 1.5 mM MgCl2, 0.2 mM ethylenediaminetetra acetic acid, 0.1% Triton X-100, 0.5 mM dithiothreitol, 20 mM glycerol phosphate, 0.1 mM Na3VO4, 2 g/ml leupeptin, 2 g/ml aprotinin, 1 mM phenylmethylsulfonyl fluoride, and a protease inhibitor cocktail tablet (Boehringer Mannheim, Mannheim, Germany). The tissue suspension was rotated at 4℃ for 10 min. Supernatants were collected and maintained at -70℃ for western blotting. Proteins from tissue were separated by SDS-PAGE using NuPAGE 4%~12% Bis-Tris gels (NP-0335Box; Invitrogen, Carlsbad, CA, USA) and then transferred to Immobilon-P (Sigma, St. Louis, MO, USA) membranes. The membranes were blocked with 5% bovine serum albumin in 1×Tris-buffered saline-0.1% Tween 20 (TBS-T) solution. The membranes were probed with primary antibody diluted 1:1,000 at 4℃ for 16 h. The blots were washed four times with 1×TBS-T buffer followed by treatment with secondary antibody for 1 h. After the secondary antibody reaction, the wells were washed four times. Proteins on the membrane were detected using the Enhanced Chemiluminescence Solution kit (Amersham, Buckinghamshire, UK). The membranes were stripped and reblotted with anti-actin antibody as a loading control (catalog #A5441; Sigma).
The primary antibodies used were anti-Nrf2 (SC-13032; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), anti-Keap-1 (60027-1-Ig; Proteintech Group, Chicago, IL, USA), and anti-Bcl-2 (#2870; Cell Signaling Technology, Danvers, MA, USA). The secondary antibodies used were horseradish peroxidase-linked anti-rabbit immunoglobulin (Ig) G (catalog #7074; Cell Signaling Technology) and anti-mouse IgG (SC-2005; Santa Cruz Biotechnology Inc.).
Subcellular fractionation
Cytoplasmic and nuclear extracts were prepared according to the instructions of the NE-PER Nuclear and Cytoplasmic Extraction kit (Pierce, Rockford, IL, USA). Subcellular fractions of normal and keloid skin tissues were used for western blot analysis.
Immunohistochemical analysis
For immunohistochemical analysis, paraffin sections (4 µm thick) were prepared and staining was performed by the avidin-biotin complex method. In brief, slides were deparaffinized in xylene and rehydrated with ethanol. The endogenous peroxidase activity was inhibited by immersion of the slides in 3% H2O2/methanol. Antigen retrieval was performed in a microwave oven for 15 min with 10 mM citrate buffer (pH 6.0). Pre-incubation took place with a blocking solution for 30 min to prevent nonspecific binding. The sections were then incubated overnight at 4℃ with the Nrf2 antibody (SC-13032, 1:500). The slides were subsequently incubated with biotinylated secondary antibody for 30 min and then with streptavidin-peroxidase, for another 30 min. The visualization of the immunoreaction was performed with 3,3'-diaminobenzidine (DBC500; ScyTek, Logan, UT, USA). Negative controls were performed in the same way, substituting the primary antibody with phosphate buffered saline. Immunohistochemical assessment was performed by measuring the intensity of labeling (-, 1+, 2+, and 3+) and the percentage of immunohistochemically stained cells (<25%, 25% ~75%, and >75%) (Table 2). The final assessment was determined by several investigators independently.
Cell culture
The normal human fibroblast and G361 cell lines served as positive controls for Nrf2 expression and Bcl-2 expression, respectively. The normal fibroblast cell line was a kind gift of Dr. Jeung Hoon Lee (Chungnam National University, Daejeon, Korea). The human malignant melanoma cell line G361 was obtained from the American Type Culture Collection (CRL 1424; Virginia, MD, USA). Cells were incubated in Dulbecco's modified eagle's medium containing 10% fetal calf serum, 100 U/ml penicillin, and 100 mg/ml streptomycin at 37℃ in a 5% CO2 incubator.
siRNA transfection
siRNA against Nrf2 and scrambled control siRNA were purchased from Invitrogen (Stealth siRNAs [Set of 3] HSS181505, HSS181506, HSS107130). The siRNA transfection was performed with Lipofectamine 2000 (catalog# 11668-019; Invitrogen), according to the manufacturer's instructions. A siRNA concentration of 80 pmol was used for a 35 mm dish of fibroblast cells.
Cell death assessment
The apoptotic cell distribution was determined using Muse Annexin V and Dead Cell kit according manufacturer's protocol (MCH100105; Merck Millipore, Billerica, MA, USA). Briefly, cells were collected by centrifugation, mixed with the Muse Annexin V and Dead Cell reagent, and analyzed using a Muse Cell Analyzer (cat. no. 0500-3115; Merck Millipore).
Cell proliferation assay
The cells were seeded in 96 well microtiter plates, followed by transfection with 10 nM Nrf2-targeting siRNA (siNrf2) or Stealth RNAi control (siC) for 24 h, 48 h, and 72 h, after which they were exposed to MTT (final 0.1 mg/ml) for an additional 4 h. The formazan crystals, formed by the reduction of MTT in living cells, were solubilized in 200 µl of dimethyl sulfoxide and the concentration was measured by spectrophotometry at 560 nm. The results were expressed as a percentage, based on the ratio of the absorbance between the treated cells and the controls (100%).
Statistical analysis
Data from the Raytest TINA software were analyzed using PASW Statistics ver. 17.0 (IBM Co., Armonk, NY, USA) and statistical significance was set at p<0.05. Data are presented as mean±standard deviation. We used the Mann-Whitney U test to compare non-normally distributed variables.
RESULTS
Protein oxidation, a marker of oxidative stress, is increased in keloid tissues in vivo
The OxyBlot analysis revealed increased oxidative stress in keloid tissues compared to normal skin tissues (Fig. 1A). Relative values for densitometric dinitrophenylhydrazone (DNP) derivatives in OxyBlot analysis were approximately 9.983×106 (9.401×106~1.093×107) in keloid and 7.644×106 (6.050×106~7.994×106) in normal skin tissues (p<0.05) (Fig. 1B). These results indicate that oxidative protein damage (DNP derivatives) is significantly increased in keloid tissues compared to normal skin.
Nrf2 is downregulated in keloid tissues compared to normal skin tissues
Western blot analysis of subcellular fractions from keloid and normal skin tissues reveal Nrf2 with the similar quantity of expression between nuclear and cytoplasmic fractions in each sample (Fig. 2A). However, the levels of Nrf2 protein are significantly lower in keloid tissues compared to normal skin tissues (Fig. 2B), whereas Keap-1 is not expressed in either keloid or control skin tissues (data not shown). The results of the Mann-Whitney U test show a median (interquartile range) of 0.3742 (0.300~0.475) and 0.5168 (0.420~0.601) for keloid and normal skin tissues, respectively (p<0.05) (Fig. 2C).
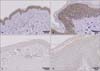
Using immunohistochemical assessment, we confirmed that Nrf2 expression is significantly downregulated in keloid tissues compared to normal skin tissues. Fig. 3 shows representative immunohistochemical staining for Nrf2 in normal skin and keloid tissues, showing significantly stronger staining of Nrf2 in normal skin compared to keloid tissues. As shown in Table 2, much less component of cells were stained with Nrf2 in keloid than normal skin tissues.
Nrf2 knockdown by transfection with siRNA induces growth-inhibiting and apoptosis-activating effects in normal fibroblasts
An increase in the rate of apoptosis was found in normal human fibroblasts after transfection with Nrf2-specific siRNA (Fig. 4). Downregulation of Nrf2 expression is observed 48 h and 72 h after Nrf2-siRNA treatment by western blot analysis (Fig. 4A). During Nrf2-siRNA treatments, phase contrast images of treated and control cells were captured. After 72 h the cell shape is deeply altered and the some of the cells appeared detached (Fig. 4B). In the apoptosis assay, three different cell populations can be detected after Annexin V-PE staining of fibroblasts: live cells group in the lower left, early apoptotic cells group in the lower right, and late apoptotic cells group in the highest right part of the panel. An increase in the apoptotic cell population is found after Nrf2-siRNA treatment (Fig. 4C). Using the MTT assay, cell proliferation is decreased in a time-dependent manner after Nrf2 knockdown in fibroblasts (Fig. 4D).
Bcl-2 is downregulated in keloid tissues compared to normal skin tissues.
Using western blot analysis, the expression of Bcl-2 is significantly lower in keloid tissues compared to normal skin tissues (Fig. 5A). The results of the Mann-Whitney U test show a median (interquartile range) of 0.266 (0.237~0.296) for keloid and 0.435 (0.359~0.490) for normal skin tissues (p<0.05) (Fig. 5B).
DISCUSSION
Keloid formation is a pathological response of wound healing to cutaneous injury in genetically susceptible individuals12. Keloids are defined as scars growing beyond the margins of the original wound that rarely regress over time13. Deposition of a dense collagenous meshwork with a rich vascular supply and increased infiltration of inflammatory cells is probably related to the proliferative and inflammatory status of keloids. There are a number of reports suggesting that oxidative stress is involved in various chronic inflammatory conditions14. In addition, oxidative damage appears to play a relevant role in the pathogenesis of various fibrotic diseases, and it has been reported that secondary production of ROS occurs after triggering inflammation due to depletion of antioxidants15. Considering these previous reports, the fibrotic and inflammatory status of keloids may be associated with ROS production. The oxidative status in keloids has been investigated in keloid fibroblasts but has not been examined in whole keloid tissues. De Felice et al.7 reported that ROS production was increased in keloid fibroblasts and suggested that this is probably involved in the pathogenesis of the disease. In our study, protein carbonyl levels, used as a marker of oxidative protein modification, were elevated in keloid tissues (Fig. 1). Our data, consistent with the findings of De Felice et al.7, suggest the presence of oxidative stress via ROS production in keloid tissues and that an impaired cellular antioxidant system may participate in this increased ROS generation.
Nrf2 comprises the main cellular antioxidant system against chemical- and radiation-induced oxidative and electrophilic stress16. Nrf2 is known to interact with actin-associated cytosolic protein, which is an inhibitor of Nrf2 (INrf2), and is also known as Kelch-like ECH-associated protein 1 (Keap1)171819202122. Keap1 functions as a substrate adaptor protein for a Cul3-Rbx1-dependent E3 ubiquitin ligase complex to ubiquitinate Nrf2, leading to its degradation and maintaining asteady state level of Nrf216. Nrf2 plays a role in cellular defense by regulating various cytoprotective proteins through Keap1-dependent or independent signaling pathways. Nrf2 is also controlled by other mechanisms that have not yet been fully elucidated. Rada et al.23 showed that degradation of Nrf2 is mediated by the Neh6 domain of mouse Nrf2 in a Keap1-independent manner. Activation of the Nrf2 defense response protects against neurodegenerative diseases, aging, diabetes, photo-oxidative stress, cardiovascular disease, inflammation, pulmonary fibrosis, acute pulmonary injury, and cancer924252627. Considering the critical role of Nrf2 against oxidative stress, a significant association between Nrf2 and increased oxidative stress in keloids may exist. However, no previous studies have investigated the role of Nrf2 expression in keloids. In our study, western blot analysis revealed that Nrf2 expression is clearly downregulated in keloid tissues compared to normal skin tissues (Fig. 2B). This result suggests that downregulation of Nrf2, the primary antioxidant system in the cell, results in the increased oxidative stress found in keloid tissues.
Ning et al.28 investigated the localization of Nrf2 in the nucleus and cytoplasm under varying concentrations of hydrogen peroxide in cultured pulmonary microvascular endothelial cells. They found that low and moderate doses of hydrogen peroxide exposure led to the nuclear accumulation of Nrf2, whereas high doses of hydrogen peroxide exposure led to nuclear exclusion of Nrf2. In our experiment, Nrf2 was found in the cytoplasmic and nuclear fractions of both normal and keloid tissues (Fig. 2A). Notably, total Nrf2 expression was lower in keloid tissues compared with that in normal tissues, while the quantity of Nrf2 expression was similar between the nucleus and cytoplasm in each sample. However, Keap1 was not detected in either normal or keloid skin tissues in the western blot (data not shown), suggesting that Nrf2 might be regulated by a Keap1-independent pathway in skin tissues. In our immunohistological analysis, much less component of cells were stained with Nrf2 in keloid than normal skin tissues (Fig. 3), which was consistent with the result of western blot analysis. These results indicate that dysregulation of ROS and Nrf2 levels may be involved in the pathogenesis of keloids.
There is growing evidence suggesting that oxidative stress resulting from ROS production is implicated in apoptosis and that excessive production of ROS results in both DNA and protein damage2930. As ROS can contribute to the induction of apoptosis, especially in inflammatory cells, the inflammatory and increased oxidative status of keloids may affect the apoptotic activity in keloid31. A few studies have attempted to explain the etiology of keloids and have reported abnormalities in the regulation of apoptosis during the wound healing process. They have suggested that pathological scarring might be a consequence of the accumulation of granulation tissue as a result of defective apoptosis mechanisms32333435. Desmoulière et al.33 have shown that the number of apoptotic cells sharply increases as the wound closes, suggesting that apoptosis is involved in the progressive transformation of granulation tissue into scar.
A few studies have reported increased apoptosis in keloid fibroblasts536. Akasaka et al.37 reported increased apoptosis in keloid fibroblast cells compared to normal fibroblasts and hypertrophic scars. Several proteins involved in apoptosis in keloid fibroblasts have also been identified. Jiménez et al.38 indicated that c-myc and rho-p21 are potent inducers of apoptosis, whereas Bcl-2, v-abl, E1B, and ras prevent activation of the apoptotic pathway in fibroblasts. Akasaka et al.36 also reported that the increased expression of caspase-3 in keloids can induce apoptosis in fibroblasts. Since ROS production can induce apoptosis, we hypothesized that Nrf2 may be involved in this process. Recent studies have suggested the possibility of an association between Nrf2 and apoptosis. In a study by Pan et al.39, an increase in apoptotic rate was found in human glioblastoma U251 cells transfected with Nrf2-specific siRNA. Consistent with these previous studies, we found that Nrf2 knockdown by siRNA produced growth-inhibiting and apoptosis- activating effects infibroblasts (Fig. 4). These data indicate that the downregulation of Nrf2 may play a role in activation of apoptotic pathways in keloid tissues.
Niture and Jaiswal40 found that Nrf2 upregulates expression of the anti-apoptotic protein Bcl-2, which leads to a reduction in cellular apoptosis in vitro. From this, we postulated that decreased expression of Nrf2 in keloid tissues may result in downregulation of Bcl-2. Indeed, we observed decreased expression of Bcl-2 in keloid tissues compared to normal skin tissue (Fig. 5). Although we did not explore the possibility of crosstalk between Nrf2 and Bcl-2, the downregulation of Bcl-2 may participate in increased apoptotic activity in keloid tissues.
These results support the previous notion that apoptosis increases in keloids and that this apoptotic dysregulation may play a crucial role in development of pathologic scarring. Given that apoptosis rates increased when Nrf2 was knocked down, we suggest that Nrf2 could be one of the central proteins involved in the pathogenesis of keloids.
To our knowledge, this is the first study to investigate the role of Nrf2 protein expression in keloid tissues. In summary, our data suggest that downregulation of Nrf2 expression in keloids could cause increased oxidative damage and lead to apoptosis in fibroblasts, which may participate in keloid pathogenesis.






 XML Download
XML Download