

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Dear Editor:
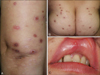
Degos disease (DD) is a rare, thrombo-occlusive vasculopathy that primarily affect the skin, gastrointestinal tract, and central nervous system1. The cutaneous lesions present as characteristic papules with porcelain-white central atrophy and an erythematous raised border. Behçet's disease (BD) is a systemic vasculitis characterized clinically by recurrent oral aphthous and genital ulcers, cutaneous lesions, and iridocyclitis/posterior uveitis2. DD can be found as an apparent idiopathic disease or as a surrogate clinical finding in some connective tissue diseases. Here, we report a patient with cutaneous manifestation of DD, associated with mucocutaneous and systemic BD lesions. A 45-year-old woman had asymptomatic erythematous papules with central, porcelain-white colored umbilication on the trunk and both extremities for 20 years, and some of the lesions had healed to leave atrophic scars (Fig. 1A, B). She also had recurrent oral and genital ulcers for 20 years (Fig. 1C). She had been treated with prednisolone, dapsone, and colchicine for 2 years, but the cutaneous lesions did not improve with BD treatments.
Histopathologic examination of a typical lesion showed a cell-poor, wedge-shaped area of dermal necrosis with vessel occlusion as well as dermal edema. Occluded vessels with thrombosis and vascular endothelial cell swelling were observed at the base of the wedge-shaped lesion, and the area was surrounded by lymphocytic infiltration (Fig. 2). Copious mucin deposition was visible in the dermis, which also showed positive staining for Alcian blue (data not shown). The laboratory findings for antinuclear antibody, lupus anticoagulant, anticardiolipin antibodies, and antiphospholipid antibodies were all negative. The results of the pathergy test were positive. The patient also had non-erosive asymmetric arthritis, and a 99m Tc-HDP 3-phase bone scan showed hot uptake. She had a scarring change on the retina due to recurrent chorioretinitis. Colonoscopy showed no specific findings for DD or BD. The patient was diagnosed with DD associated with BD. The cutaneous lesions, which were not responsive to BD treatment, improved after 4 months of treatment with aspirin and dipyridamole.
While the etiology of DD is unknown, evidence suggests a primary vaso-occlusive process. Vascular coagulopathy, endothelial cell damage, and an autoimmune process are considered the major pathogenic mechanisms3,4. In BD, an immune-mediated occlusive vasculitis is found in most sites of the active disease, and the endothelium appears to be the primary target5. The cutaneous lesions of the present patient were characteristic of DD and did not respond to BD treatments. This indicates that, while DD can be associated with BD, it is a distinct disease entity to BD.
To the best of our knowledge, although Sweet's syndrome and pyoderma gangrenosum in BD have been reported in association with BD, our patient represents the first reported case of DD associated with BD. With the underlying vaso-occlusive process of both DD and BD, it is reasonable for an association between DD and BD to exist. Whether this patient had the same underlying mechanisms or the findings were coincidental is yet to be determined. We suggest that DD and BD are distinct disease entities but that DD can be associated with BD.

 XML Download
XML Download