

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ashy dermatosis is an idiopathic ash-colored macular hyperpigmentation disorder in young adults. It is usually slowly progressive and leaves permanent discoloration. Poikiloderma vasculare atrophicans (PVA) is poikilodermatous mycosis fungoides (MF) characterized by poikiloderma, hyper- and hypopigmentation, atrophy, and typical telangiectasia on the trunk. PVA with an epidermotropism of CD4-CD8+ atypical T cells responds well to phototherapy and has a good overall prognosis. Herein, we present a case of PVA showing features of ashy dermatosis in the beginning.
CASE REPORT
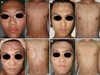
In 2001, a 14-year-old male patient presented with asymptomatic brownish black-pigmented reticulated patches over the whole body, particularly on the neck, axillary, and antecubital areas, for an unknown period (Fig. 1A). The results of laboratory studies were all within normal limits or were negative. The histologic examination showed increased number of melanophages in the papillary dermis with focal hydropic changes in the basement membrane zone (Fig. 2A). Ashy dermatosis was considered a possible diagnosis, but the patient was lost to follow-up.
In 2005, the patient revisited with scaly increased darkening and lichenification on the previous lesions in the absence of any interim treatment (Fig. 1B). The skin biopsy revealed interface dermatitis with band-like infiltration in the upper dermis at the epidermo-dermal junction with exocytosis and necrotic keratinocytes (Fig. 2B). The patient was diagnosed with active-phase ashy dermatosis, and treatment with regular follow-up was recommended. However, he refused treatment at that time.
In January 2011, the patient visited again with a prominent poikilodermatous change on the previous skin lesions (Fig. 1C). The physical examination and laboratory test results were all within normal limits or were negative. A skin biopsy performed on the flank area showed epidermal atrophy and lymphocytic infiltration in the upper dermis with epidermotropism of atypical lymphocytes (Fig. 2C). A CD4~CD8 double stain revealed a 1 : 1 ratio of CD4 cells to CD8 cells, and the atypical T cells were mainly CD4-CD8+ (Fig. 2C'). A retrospective CD4-CD8 double staining on the previous biopsy specimen also revealed a 1 : 1 ratio of CD4 cells to CD8 cells and mild epidermotropism of CD4-CD8+ lymphocytes (Fig. 2A', B'). The results of the T-cell receptor (TCR)γ gene rearrangement analysis were positive for clonality in the affected skin lesions. Based on the aforementioned findings, PVA (stage IB) was diagnosed. We treated the generalized skin involvement with topical steroids and narrow-band ultraviolet B (NB-UVB) phototherapy. In March 2012 after 1 year of treatment, the poikilodermatous lesions had disappeared and atypical lymphocytes were not observed on a follow-up skin biopsy (Fig. 1D, 2D, 2D'). The results of another TCRγ gene rearrangement analysis were also negative. Currently, the patient undergoes regular observation after 6 months of biweekly NB-UVB phototherapy. No new suspicious skin lesions or palpable lymph nodes have been observed thus far.
DISCUSSION
PVA was first introduced by Jacobi in 1908 to describe a condition that was later regarded as a dermatomyositis1. Recent review articles have reported PVA as poikilodermatous MF characterized by mottled pigmentation (hyperand hypopigmentation), atrophy, and typical telangiectasia involving the major flexural areas and the trunk2,3. Abbott et al.4 identified 49 patients who predominately had poikilodermatous MF. There was a slight predominance of male patients 1.6 : 1 (30 of 49) with a median age of 44 years4,5. Most patients (43 of 49) had an early disease stage (≤IIA) at diagnosis, and no patients had stage IV disease at presentation4. The histopathologic features, in addition to those associated with classic MF, included atypical T lymphocyte infiltration and epidermal atrophy in the papillary dermis and epidermis, pigmentary incontinence, and telangiectatic vessels6. In PVA, a predominant CD4-CD8+ phenotype was detected on immunohistochemistry (15/40; 38%) compared with classic MF, which had a CD8+ phenotype in just 6.5% of cases according to previous studies4,7. Our patient also revealed mild epidermotropism of the CD4-CD8+ lymphocytes in retrospective CD4-CD8 double staining accompanied by similar clinical features of ashy dermatosis in the beginning (Fig. 2A', B'). Thus, careful pathologic evaluation can lead to early diagnosis of the disease or at least a differential diagnosis. Patients with PVA respond well to phototherapy, which is the most commonly used first-line therapy, and they have a good overall prognosis4. To the best of our knowledge, four cases involving a similar presentation were reported in the Korean literature as parapsoriasis variegata in 1979, as poikilodermatous MF in 1999, and as PVA in 1999 and 20118,9,10,11.
Ashy dermatosis or erythema dyschromicum perstans presents as asymptomatic confluent ashy colored macular hyperpigmentation that is idiopathic, slowly progressive, and permanently discoloring12, and usually appears in young adults. The trunk and proximal extremities are more commonly involved, followed by the neck and face8. The histopathologic features show dermal perivascular lymphocytic infiltration, vacuolization of the basal cell layer, occasional colloid bodies, and incontinence of pigment. Since there is no effective treatment, a number of different therapeutic approaches have been attempted.
CD4-CD8+ MF is a rare form of MF (fewer than 5%), which is characterized by chronic epidermotropism of predominately CD8+ atypical T cells and often hyperpigmentation and poikiloderma. Nikolaou et al.7 presented a series of seven cases of CD8+ cytotoxic variant MF and reported that CD8+ MF follows an indolent course, responds well to phototherapy, and has a benign prognosis. Based on these findings, they suggested that the CD8+ immunophenotype may represent a marker of mild biological behavior7. Our patient also showed a CD4-CD8+ immunophenotype, which may explain the benign clinical course over a 10-year period. Thus, if a patient shows long-standing and progressive hyperpigmentary skin changes, periodic follow-up and repeated skin biopsies are recommended to determine the underlying condition.
In conclusion, our case demonstrates four important points. First, the clinical and pathologic findings of early stage PVA can mimic ashy dermatosis. Therefore, if a patient shows long-standing and progressive hyperpigmentary skin changes, periodic follow-up and repeated skin biopsies are recommended to determine the underlying condition. Second, we confirmed that PVA or poikilodermatous MF is a rare variant of MF, which has a benign clinical course and presents with the CD8+ phenotype more frequently4. Third, we also confirmed that CD8+ MF presents with special clinical features such as hyperpigmentation and poikiloderma. Finally, this case had an indolent, benign course, which corroborates the finding that the CD8+ immunophenotype of MF may represent a marker of mild biologic behavior4.

 XML Download
XML Download