

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Systemic sclerosis (SSc) is a connective tissue disease characterized by excessive extracellular matrix (ECM) deposition in the skin and visceral organs, with an autoimmune background1. Although the pathogenesis of SSc remains unknown, three major types of abnormalities-collagen accumulation, vascular injury, and immune activation-are considered as the main features of the disease2,3,4. Collagen accumulation mainly results in fibrosis in the skin and lungs. Vascular injury results in Raynaud's phenomenon, digital ulcers, scleroderma renal crisis, and pulmonary hypertension. Immune activation is characterized by autoantibody production, lymphocyte activation, and release of various cytokines. The presence of autoantibodies is a central feature in immune activation associated with SSc, because antinuclear antibody (Ab) has been detected in >90% of the patients5. SSc patients have autoantibodies that react to various intracellular components, such as DNA topoisomerase I (topo I), centromeres, RNA polymerases, U1RNP, U3RNP, Th/To, and histones5. In addition, the cytokines that are released by lymphocytes infiltrating the affected tissue may cause vascular injury and collagen production. However, it has not been possible to unify the three major types of abnormalities into one hypothesis.
SSc is a heterogeneous disorder since it includes a very broad spectrum of clinical manifestations. Therefore, an appropriate classification of disease subsets is necessary to evaluate clinical manifestations, predict prognosis, and select appropriate treatments for each patient. According to the most widely accepted LeRoy's classification1, SSc has been classified into diffuse cutaneous SSc (dcSSc) and limited cutaneous SSc (lcSSc). Patients with skin sclerosis proximal to the elbow are considered to have dcSSc, while patients with skin sclerosis distal to the elbow have lcSSc. The specificities of the autoantibodies closely correlate with the clinical manifestations. For example, anti-topo I Ab and anti-RNA polymerase Ab are associated with dcSSc, while anti-centromere Ab and anti-Th/To Ab are generally detected in lcSSc. These observations suggest that autoantibodies are closely linked to the pathogenesis of SSc. Nonetheless, these autoantibodies are not likely to have a direct pathogenic role in SSc, because most SSc-related autoantigens are intracellular components. Autoantibodies are usually not internalized into the cell. A recent study has shown that anti-topo I Ab could directly bind to the cell surface of fibroblasts6. In addition, mice immunized with topo I showed skin sclerosis and lung fibrosis7. However, the pathological relevance of anti-topo I Ab remains unclear. Collectively, at the minimum, the presence of autoantibodies demonstrated that abnormal B cell activation exists in SSc.
Recent studies have indicated that B cells played more varied fundamental roles in regulating immune responses8,9 than what had previously been appreciated (Fig. 1). These roles include antigen presentation, cytokine production, lymphoid organogenesis, T cell differentiation, and influence on dendritic cell and macrophage function. Consistently, not only autoantibody production, but also abnormalities in other B cell functions, could lead to the induction or development of autoimmune disorders. SSc patients have polyclonal B cell hyperactivity and hyper-γ-globulinemia in addition to undergoing autoantibody production10. Chronic B cell activation is likely to be critical for the development and progression of the disease. In this review, we focus on how altered B cell function may be linked to tissue fibrosis in SSc through autoimmuity.
RESPONSE REGULATORS OF B CELL SIGNALING
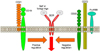
B cells respond to many types of stimulation and regulate the negative selection in bone marrow, the generation of humoral immune responses in the peripheral lymphoid organs, and the establishment and maintenance of tolerance and memory. The outcome of these B cell responses is determined by signaling thresholds through the B cell antigen receptor (BCR) complex11. The signaling thresholds are regulated by response regulators, which are an array of cell surface molecules or cytoplasmic signal transduction molecules that augment or diminish BCR signals during B cell development as well as during responses to self and foreign antigens (Fig. 2)11,12,13. Thus, "response regulators" is a useful concept in understanding how autoimmunity is induced as well as how normal humoral immune responses are facilitated.
Recent analyses using mice lacking or overexpressing certain molecules have identified many molecules that act as response regulators (Fig. 2). To easily understand the function of each molecule, it has been convenient to categorize molecules as being either positive or negative response regulators of B cell function11,12,13. CD19, CD21 (complement receptor type 2, CR2), and CD45 have been identified as a group of positive response regulators, which augment signals through the BCR complex, whereas CD22, CD72, FcγRIIB, Src homology 2 domain-containing tyrosine phosphatase-1 (SHP-1), and Src homology 2 domain-containing inositol polyphosphate 5'-phosphatase have been grouped as negative response regulators that diminish BCR signals11,12,13,14,15,16. For example, transgenic mice that overexpress CD19 lost tolerance for autoantigen and generated autoantibodies spontaneously17,18. Mice lacking CD22 had chronically activated B cells with various spontaneous autoantibody production19,20. Motheaten viable (mev/mev) mice with SHP-1 mutations produced elevated levels of spontaneous autoantibodies and hyper-γ-globulinemia, and induced tissue deposition of immune complexes21. These data suggest that the response of B cells to foreign or self-antigens is controlled in part by interactions between positive and negative response regulators. Therefore, altered function or expression of these molecules can influence the susceptibility to autoimmunity. Taken together, abnormal regulation of the function and expression of response regulators may result in autoantibody production. Among the response regulators, CD19, which is a critical cell-surface signal transduction molecule of B cells, is one of the most potent positive regulators.
CD19 AS A POSITIVE RESPONSE REGULATOR OF B CELL SIGNALING
CD19 expression is restricted to B lineage cells and follicular dendritic cells, which are antigen-presenting cells located in the murine spleen22. CD19 is a 95,000 Mr glycoprotein of the immunoglobulin (Ig) superfamily expressed from early pre-B cells until plasma cell differentiation23. On B cells, CD19 is associated with CD21, a receptor for complement C3 cleavage fragments and Epstein-Bar virus. CD19 expression is tightly regulated during the B cell activation process, suggesting that intrinsic CD19 expression levels may determine a predisposition to autoimmunity. CD19 has an extracellular region containing two C2-type Ig-like domains and a cytoplasmic region of ~240 amino acids with 9 conserved tyrosine residues24. Lyn, a Src-family protein tyrosine kinase member, is the dominant kinase that phosphorylates CD19 upon stimulation. Once tyrosyl-phosphorylated, CD19 serves as a membrane-bound adaptor protein for Src homology 2-containing signaling molecules such as Lyn, Vav, and phosphatidylinositol 3-kinase, which further mediate downstream activation cascades.
Recently, in vivo CD19 function was assessed using CD19-deficient mice and CD19-transgenic mice, which overexpressed CD19 by 300%18,25,26,27,28,29. CD19-deficient B cells exhibited lower proliferation than wild type B cells in response to various transmembrane signals, while B cells from CD19-transgenic mice showed augmented proliferation25,28. Serum Ig levels were spontaneously increased in CD19-transgenic mice, while they were decreased in CD19-deficient mice. Moreover, serum levels of autoantibodies including anti-topo I, anti-DNA, and anti-histone Abs were decreased in CD19-deficient mice, whereas those in CD19-transgenic mice were increased18. Thus, CD19 expression in B cells showed a close positive correlation with the production of autoantibodies. Furthermore, analysis using CD19-transgenic mice with autoreactive B cells has revealed that CD19 overexpression disrupted peripheral tolerance in B cells and thereby induced autoantibody production and autoimmunity17. These results suggest that CD19 expression levels regulate autoantibody production by augmenting B cell signaling. Interestingly, B cells stimulated with anti-IgM Ab or lipopolysaccharides increased the expression of I-A, a major histocompatibility complex class II molecule that is a marker of B cell activation, while CD19 expression was not affected by B cell activation18. Thus, CD19 expression is tightly regulated during the B cell activation process, suggesting that overexpression of CD19 can lead to autoimmunity.
CD19 EXPRESSION IN B CELLS FROM SSC PATIENTS
As observed in the flow cytometric analyses of blood from SSc patients, the surface density of CD19 in SSc B cells was significantly higher by ~20% than that in healthy individuals30. CD19 overexpression was detected in both naive B cells and memory B cells from SSc patients31. Furthermore, CD21 expression was higher in SSc patients30. In contrast, CD40 and CD20 levels were normal. Although CD19 expression levels were higher on B cells from SSc patients, the increase in CD19 expression was small (~20%), and it remains unknown whether this small increase is related to autoimmunity. Therefore, the pathogenic significance of the 20% increase in CD19 expression was assessed by generating transgenic mice that overexpressed CD19 to a similar extent as human SSc did29,31. These transgenic mice that overexpressed CD19 by 20% had significantly elevated levels of various autoantibodies, including SSc-specific anti-topo I Ab as well as anti-DNA Ab, anti-histone Ab, and rheumatoid factor31,32. These results suggest that the small increase in CD19 expression observed in human SSc may be sufficient to induce autoantibody production. However, the transgenic mice did not develop fibrosis in the skin and visceral organs. Thus, it is still unclear whether CD19 overexpression and autoantibody production are related to disease development and progression in SSc.
In addition to significant autoantibody production, hyper-γ-globulinemia and polyclonal B cell hyperactivity were detected in SSc patients33,34. Recent analysis of gene expression using DNA microarrays has revealed an up-regulation of the expression of genes related to B cells10. These observations suggest the presence of intrinsic B cell abnormalities in SSc. To assess whether intrinsic B cell abnormalities exist in SSc, phenotypic and functional abnormalities of blood B cell subsets were assessed8,35. Importantly, in patients with SSc, total blood B cells were expanded. In addition, peripheral B cell homeostasis and subsets were disturbed in SSc. Although memory B cells and plasmablasts/early plasma cells diminished, naive B cells expanded. Furthermore, memory B cells from SSc patients showed increased expressions of CD80 and CD86. Since it is generally accepted that B cell activation is required to up-regulate the expression of both CD80 and CD86, which are crucial co-stimulatory molecules of B cells, this finding indicates that memory SSc B cells are chronically activated in vivo, possibly because of CD19 overexpression. Interestingly, CD95 (Fas) expression, which is up-regulated following B cell activation, was also increased in SSc memory B cells. The increased Fas expression induced marked sensitivity to Fas-mediated apoptosis36. This enhanced sensitivity to spontaneous apoptosis of SSc memory B cells may result in a diminished number of these cells in the blood. In addition, it is possible that the continuous loss of memory B cells and plasmablasts/early plasma cells increases the production of naive B cells in bone marrow to maintain B cell homeostasis. Remarkably, although the number of memory B cells decreased in SSc patients, SSc memory B cells have an enhanced ability to produce IgG, resulting in hyper-γ-globulinemia and possibly autoantibody production. Thus, SSc patients have distinct abnormalities in B cell compartments characterized by expanded naive B cells and activated memory B cells. Furthermore, CD19 overexpression in memory SSc B cells may be related to their hyper-reactivity, since memory B cells as well as naive B cells from SSc patients overexpress CD19.
B CELL ABNORMALITIES IN TIGHT-SKIN/+ MICE
The tight-skin (TSK)/+ mouse is a genetic model for human SSc and was originally identified as a spontaneous mutation that results in increased synthesis and accumulation of ECM proteins including collagen. A tandem duplication within the fibrillin 1 gene was suggested to cause the TSK phenotype37. Fibrillin 1 is a major structural protein of widely distributed microfibrils. Although homozygous mutation results in embryo lethality, heterozygous mice survive and develop cutaneous hyperplasia, pulmonary emphysema, and cardiac hypertrophy. TSK/+ mice produce autoantibodies against SSc-specific target autoantigens including topo I, fibrillin 1, and RNA polymerase I, while the role of a fibrillin 1 gene in the genesis of tissue fibrosis and autoimmunity remains unsolved.
The symptoms of TSK/+ mice are regulated not only by fibrillin 1 mutation but also by multiple factors. The transgenic mice overexpressing the fibrillin 1 gene developed cutaneous hyperplasia, but not pulmonary emphysema and myocardial hypertrophy38. CD4 deficiency in TSK/+ mice decreased cutaneous hyperplasia but did not affect lung emphysema or anti-topo I Ab levels39. In addition, interleukin-4 deficiency did not cause cutaneous hyperplasia but resulted in pulmonary emphysema40. Furthermore, tumor growth factor-β1 promoter polymorphisms have been reported41. These results suggest that abnormal immune functions contribute to the phenotype of TSK/+ mice.
B cells from TSK/+ mice also have abnormal phenotype32,42,43. Unlike in human SSc, CD19 overexpression was not detected in TSK/+ B cells. However, constitutive CD19 tyrosine phosphorylation was upregulated in TSK/+ B cells. Furthermore, intracellular Ca responses generated by CD19 ligation were increased in TSK/+ B cells. Collectively, the CD19 signaling pathway appeared to be constitutively activated in TSK/+ B cells, resulting in increased early B cell responses mediated by CD19. Consistent with spontaneously enhanced CD19 signaling, TSK/+ B cells showed a phenotype of chronic B cell activation. Furthermore, in CD19-deficient TSK/+ mice, chronic B cell activation was inhibited. Thus, TSK/+ B cells exhibited the constitutively activated phenotype because of abnormal basal signaling thresholds.
To assess the effect of the TSK/+ mutation on B cell responsiveness, serum Ig levels were evaluated in TSK/+ mice42. TSK/+ mice exhibited increased serum Ig levels, which were reduced by CD19 deficiency, indicating that hyper-γ-globulinemia in TSK/+ mice is dependent on CD19 function. In addition, TSK/+ mice had significantly elevated serum levels of autoantibodies including anti-topo I Ab. Remarkably, CD19 deficiency in TSK/+ mice completely abrogated the production of autoantibodies. Therefore, the loss of CD19 expression dramatically inhibited autoantibody production in TSK/+ mice. Furthermore, CD19 deficiency in TSK/+ mice showed a 40% reduction of cutaneous hyperplasia. Therefore, B cells contributed to skin fibrosis in TSK/+ mice by augmenting a CD19 dependent pathway.
Alternatively, abnormalities of other response regulators might affect CD19 signaling. CD22 specifically reduced TSK/+ B cell activation32. CD22 has immunoreceptor tyrosine-based inhibitory motifs that activate SHP-1 when phosphorylated. CD22 acts as an inhibitory signaling molecule, and a major target of the CD22/SHP-1 pathway is CD19. Therefore, disrupted regulation of CD22 in TSK/+ B cells may result in abnormal activation of downstream signal transduction molecules including CD19.
B CELLS AND BLEOMYCIN-INDUCED SSC MOUSE MODELS
Recently, a new mouse model of SSc has been established using bleomycin (BLM) treatment44,45. BLM, whose side effects include pulmonary fibrosis or scleroderma-like conditions, is an antibiotic widely used for cancer treatment. To date, several studies have shown that B cells play a critical role in the development of fibrosis and autoantibody production in TSK/+ mice8,35,46,47. However, this hypothesis remains unproven since there are important differences between the TSK/+ mouse model and human SSc. First, skin fibrosis in TSK/+ mice is located in the subcutaneous connective tissue layer, which does not exist in humans, while fibrosis in human SSc occurs in the dermis. Second, cell inflammation in the dermis, which regulates skin fibrosis by producing cytokines in human SSc, is very modest in TSK/+ mice. Third, TSK/+ mice exhibit lung emphysema, whereas human SSc is associated with lung fibrosis. Finally, human SSc is not a genetic disorder as exhibited in TSK/+ mice, because SSc occurs in only 1.6% of families with previous occurrence of SSc48. In this regard, the subcutaneous injection of BLM induces fibrosis in the dermis and lung, autoantibody production, and dermal inflammatory cell infiltration, mimicking more closely the features of human SSc than those of TSK/+ mice44,45.
To further assess the role of CD19 in systemic autoimmunity and tissue fibrosis, CD19 deficiency was investigated in the BLM-induced SSc mouse models49. CD19 deficiency inhibited the development of skin and lung fibrosis, hyper-γ-globulinemia, and autoantibody production in the BLM-induced SSc mouse models. This reduced dermal fibrosis by CD19 deficiency was associated with the decreased dermal infiltration of macrophages, mast cells, T cells, and B cells, suggesting that B cell activation contributes to downstream inflammatory infiltration of other immune cells. Recently, it was found that toll-like receptors (TLRs) and their ligands contributed to inflammatory responses, including autoimmune diseases, as well as host defense by innate immunity50. For example, lipopolysaccharides, a major component of gram-negative bacteria, are an exogenous ligand for TLR4 that induce B cell activation. Previous studies have identified various endogenous ligands for TLR4, such as hyaluronan and other ECM proteins, which regulate inflammatory responses51,52,53. Importantly, BLM treatment enhanced hyaluronan production in the skin, lung, and sera. Consecutively, hyaluronan stimulated B cells to produce various cytokines mainly via TLR4. Remarkably, CD19 deficiency generally inhibited hyaluronan-induced cytokine production by B cells49. These results suggest that ECM components, especially hyaluronan, cooperatively regulate fibrosis by inducing cytokine production by B cells, which is largely dependent on CD19 signaling.
BLM treatment induces the production of autoantibodies, especially SSc-specific anti-topo I Ab, and hyper-γ-globulinemia, both of which are central features of human SSc. It has been hypothesized that immune responses to autoantigens are induced by cryptic self-epitopes that are generated by modification of the self-antigens during apoptosis54,55. In addition, topo I was clustered and concentrated in the surface blebs (apoptotic bodies) of apoptotic cells, which may result in the antigen presentation of the cryptic epitopes. Importantly, apoptosis was detected in endothelial cells of early inflammatory lesions in patients with SSc56. Similarly, in the BLM-induced SSc model, apoptosis was prominently detected in the skin57. Collectively, apoptosis induced by BLM and enhanced TLR signaling of B cells by hyaluronan may induce autoantibody production. Furthermore, the finding that a loss of CD19 eliminated autoantibody production suggests that CD19 regulates autoantibody production in response to BLM treatment possibly by altering TLR4 signaling.
CONCLUSION
B cells as a potential target in SSc therapy
Recently, the role of B cells has been highlighted in human SSs. Similar to B cells in SSc patients, TSK/+ B cells exhibit augmented CD19 signaling. This enhanced CD19 expression/function may result in autoantibody production through a breakdown of B cell peripheral tolerance. Chronic B cell activation may result in the development of skin fibrosis through continuous production of cytokines in TSK/+ mice. On the other hand, in the BLM-induced SSc mouse models, BLM induces autoantigen presentation during tissue injury and endogenous TLR4 ligand production, which results in autoantibody production and fibrosis. Recent studies have revealed that BLM also induces the production of reactive oxygen species (ROS)58,59. In the BLM-induced lung fibrosis model, inflammatory cells and lung epithelial cells were responsible for ROS58. Mice deficient in ROS production showed a reduction in BLM-induced lung fibrosis, suggesting that ROS generated by BLM induced tissue damage and fibrosis59. In addition, a free radical scavenger, edaravon, inhibited skin and lung fibrosis in the BLM-induced SSc mouse models60. Similarly, many previous studies have confirmed that human SSc patients showed enhanced ROS production, which was probably due to ischemia and reperfusion injury following Raynaud's phenomenon, an initial clinical manifestation in human SSc61,62. Thus, enhanced ROS production, which is a common feature in human SSc and the BLM-induced SSc model, plays a role as an initiator of tissue injury and fibrosis58,59,61. Furthermore, ROS are responsible for hyaluronan degradation, leading to the production of hyaluronan fragments that induce inflammatory responses63. Likewise, hyaluronan levels were increased in plasma or sera from human patients with SSc, especially early SSc, and were correlated closely with the severity and progression of the disease as well as the degree of visceral involvement53,64,65. Collectively, we have proposed the following hypothesis (Fig. 3): enhanced ROS production, which is induced by BLM in the BLM-induced SSc mouse models and Raynaud's phenomenon in human SSc, could initiate tissue damage, leading to generation of hyaluronan fragments, which induce B cell activation mainly via CD19 and TLR4. Moreover, the abnormal B cell activation may be associated with autoantibody production and augmented cytokine production that may result in fibrosis. These findings suggest that CD19 and TLR4 signaling of B cells could be potential therapeutic targets in the treatment of human SSc.

 XML Download
XML Download