

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Epidermolysis bullosa simplex (EBS) is a chronic vesicular disorder with characteristic manifestations, from birth to infancy, of intraepidermal vesicle and milia formation on the hand, elbow, or knee due to minimal trauma. It is a genetic disorder that is caused by a dominant-negative mutation in either the keratin 5 (KRT5) or the keratin 14 (KRT14) gene. EBS is sub-categorized by its clinical manifestation into the systemic (Koebner), localized (Weber-Cockayne), and herpetiform (Dowling-Meara)1 types. The localized type of EBS is the mildest form of the subtypes that involves easy development of vesicles on the palms and soles from minimal mechanical trauma. According to molecular genetic studies of EBS, there are mutations in KRT5 and KRT14, which contribute to skeletons on hemidesmosome in keratinocytes located in the basal layer near the dermo-epidermal junction. Mutations in each subtype of EBS vary in location and severity2,3.
In our case, a 5-year-old Korean boy, who, from infancy, developed blisters and erosions on his palms and soles, visited Keimyung University Dongsan Medical Center. Through physical examination, discussion of family history, skin biopsy, and mutation analysis, we identified a novel mutation of thymine-to-cytosine transition at codon 608 in KRT5. From these results, the patient was diagnosed with Weber-Cockayne type EBS.
CASE REPORT
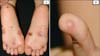
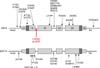
A 5-year-old Korean boy with blisters and erosions on the palms and soles visited our clinic. The symptoms had been recurring since infancy from minimal mechanical trauma. Upon physical examination, several bullae vesicles with crusts and erosions on both soles, and a vesicle on the fingertip were observed on the patient. The oral mucosa appeared to be asymptomatic (Fig. 1). All affected lesions were limited to trauma-prone sites. A skin biopsy revealed a large intraepidermal vesicle in the suprabasal layer with scanty inflammation of the dermal portion (Fig. 2). These phenomena were recorded in the family history and presented in a pedigree with his younger sister and his father (Fig. 3A). Mutational analysis of the patient and parental DNA showed a thymine-to-cytosine transition mutation in codon 608 of exon 2 in the KRT5 gene in the patient and his father only (Fig. 3B). Identical digestion patterns were observed for the patient and his father in an RFLP analysis with Pst I (Fig. 3C), but the patterns differed for his mother and the control group. These results indicate that the mutation was inherited from his father. The patient was treated with systemic and topical antibiotics as supportive treatment during the diagnosis as well as during regular follow-up appointments (Fig. 4).
DISCUSSION
Epidermolysis bullosa, a rare genetic disorder, is categorized into EBS, junctional epidermolysis bullosa, and dystrophic epidermolysis bullosa, depending on the extent of vesicle formation and its clinical manifestations. EBS is caused by mutations in the genes coding for KRT5, a member of the type II group keratin located in chromosome 17, and KRT14, a member of the type I group keratin located in chromosome 12. An "α-helical rod" domain, composed of 4 subdomains, plays a key role in the pairwise conjugation of keratin, which is needed for structural support against friction4,5. Mutations in this domain can cause severe problems in the assembly of keratin filaments6. Hence, the sites of the mutations are important for determining the clinical repercussions of mutant proteins. For example, mutations in the ends of the subdomains result in the herpetiform type-the most severe clinical form. In contrast, mutations in the central portions of the subdomains result in a less severe form, the Dowling-Meara type EBS. The mildest form of EBS, the localized type, results from mutations in the head, tail, or non-helical portions, including the linker area7. In our case, the location of the neutral L608P mutation was in a non-helical portion (linker area) and resulted in a localized type EBS.
While the location of the KRT5 or KRT14 mutation can affect the severity of EBS, the polarity of the substituted amino acid residue is also important. Even mutations within the same region can lead to various clinical symptoms, depending on the polarity of the amino acid. Similarly, there can be severe clinical findings in case of a change in the acid/base properties of a residue because of the destabilizing effects exerted onto the protein structure8. Predictably, if the polarity of amino acid does not change, such as neutral to neutral, only mild clinical findings are observed. For example, in our study, a neutral leucine was mutated to a neutral proline. In conclusion, we can presume that the localized/Weber-Cockayne type EBS in this patient was induced by the neutral substitution of a leucine residue for a proline residue in the non-helical area (linker region) of keratin 5.


 XML Download
XML Download