

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Neu et al.1 and Laxova et al.2 independently reported siblings with a lethal syndrome of marked intrauterine growth retardation, microcephaly, flexion deformities of the limbs, bizarre facial features, ichthyosis and various other anomalies, including slanted forehead, flat nose, short neck, widely spaced nipples, absence of corpus callosum and patent foramen ovale. In 1979, Lazjuk et al.3 described a stillborn female with similar findings and proposed that their case and those previously described by Neu et al.1 and Laxova et al.2 represented a new, distinct genetic syndrome; hence, the eponym Neu-Laxova syndrome (NLS) was coined.
To date, approximately 72 cases of NLS have been reported in the medical literature. Parental consanguinity has been reported in 42% of the cases. Maternal history of spontaneous abortions and polyhydramnios is common and affected infants typically have a normal karyotype4.
Although NLS is generally a lethal condition, cases of patients who lived beyond 6 months and 10 months of age have been reported in the literature5,6. In this article, we present a 3-year-old boy who was born with collodion membrane, facial dysmorphic features, limb anomalies, genital hypoplasia and pachygyria. We believe that the clinical findings in this patient may represent a mild form of NLS or a new genetic entity.
CASE REPORT
A male newborn born at 37 week 4 days gestation was referred to us with the presence of a collodion membrane and multiple congenital anomalies. The mother, a healthy 38-year-old gravida 4 para 3 woman, was married to her maternal first cousin, a healthy 41-year-old man. The baby was born by spontaneous vaginal delivery and his birth weight, height and head circumference were 3,930 g, 50 cm and 34 cm, respectively. There was no history of complications during pregnancy or maternal drug exposure. The first two pregnancies had been uneventful and the mother had given birth to two healthy full-term female babies who are currently 21 and 17 years old. As she was having treatment for nephrolithiasis, the third pregnancy had been terminated by curettage. The patient's family history was otherwise unremarkable.
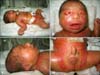
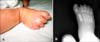
On physical examination, a parchment paper-like membrane covering the entire skin, generalized erythema and edema were noted (Fig. 1A). The infant also had a slanted forehead, hypertelorism, ectropion, broad and depressed nasal root, eclabium, micrognathia (Fig. 1B) and low-set and malformed ears (Fig. 1C). Additional abnormal clinical findings included hypoplastic testes (Fig. 1D), bilateral cryptorchidism, flexion contractures of the lower extremities, distal hypoplasia of the fingers and toes and syndactyly of the second and third toes of the right foot (Fig. 2A).
The results of the laboratory tests, including complete blood count, liver and kidney function tests, along with the measurement of serum levels of electrolytes were all within normal limits. Detailed metabolic screening tests, including tandem-mass analysis, were normal.
The chest radiograph, abdominal ultrasonography and echocardiographic examination showed no abnormal findings. Radiographic examination of the skeletal system revealed hypoplasia of distal phalanges of feet, and syndactyly of the second and third toes of the right foot (Fig. 2B). Brain magnetic resonance imaging (MRI) demonstrated pachygyria of bilateral frontal lobes and normal corpus callosum. Orbital MRI was normal. Chromosome analysis performed on a peripheral blood sample showed a normal karyotype (46,XY).
Histopathological examination of a lesional skin biopsy revealed hyperkeratosis and minimal acanthosis of the epidermis. The granular layer was normal. The dermis showed congested vascular sructures. These histopathological findings were consistent with ichthyosis.
A molecular genetic analysis of DNA extracted from a peripheral blood sample was performed in order to differentiate Hutchinson-Gilford progeria syndrome (OMIM 176670) and restrictive dermopathy (RD) (OMIM 275-210). The lamin A (LMNA) sequence showed 2 common heterozygous polymorphisms: c.1698C>T in exon 10 and c.2095C>A in exon 12. The FACE1 (ZMPSTE24) sequence was wild type.
The patient was started on supportive treatment. He was placed in a humidified incubator to prevent hypothermia and dehydration. The fluid and electrolyte balance was maintained and skin emollients containing petrolatum was used. The management of ectropion was conducted by an ophtalmologist.
He had no major health problems during the next 8 years of follow-up. An operation was performed by a pediatric surgeon for bilateral cryptorchidism. When the patient was 3 years old, the physical examination revealed ichthyosis, limb anomalies (distal hypoplasia of the fingers and toes, bilateral syndactyly of the second and third toes, flexion contractures of the distal phalanges on the index fingers) and facial dysmorphic features (broad and depressed nasal root, micrognathia, low-set and mildly dysmorphic helices). The patient demonstrated a mild mental/motor retardation, which was confirmed by the results of the Stanford-Binet test. His weight, height and head circumference were 14.8 kg (between the 25th and 50th percentiles for age), 92 cm (between the 10th and 25th percentiles for age) and 49 cm (within the normal standard deviation), respectively. The skeletal survey was all normal except for the hypoplastic distal phalanges of the toes and fingers.
DISCUSSION
NLS is a rare, lethal, autosomal recessive disorder1-4. Clinical features of this syndrome include intrauterine growth retardation, central nervous system anomalies (microcephaly, lissencephaly, hypoplastic cerebellum, agenesis of corpus callosum, microgyria), skin findings (ichthyosis, edema, collodion baby, harlequin fetus), facial dysmorphic features (slanted forehead, hypertelorism, ectropion, flat/abnormal nose, lowset/malformed ears, eclabium, micrognathia, cleft lip/palate), limb anomalies (flexion contractures, deformity of digits, deformity of limbs, syndactyly of fingers and toes, rockerbottom feet, scoliosis) and genital hypoplasia4. Primary abnormalities of the muscles, arteries, nerves and bones, pulmoner hypoplasia, cardiac and renal anomalies have also been reported7.
Although diagnosis of NLS is based on the constellation of clinical findings, no formal diagnostic criteria have been proposed. However, a number of distinguishing features have been suggested; namely, primary features (intrauterine growth retardation, neonatal lethality, microcephaly, short neck, edema, ichthyotic skin lesions) and secondary (deformation) features (slanted forehead, flat nose and nasal bridge, rocker-bottom feet, swollen hands and feet, interdigital webs)8. It is noted that the combination of the central nervous system and limb anomalies with severe subcutaneous edema observed in the prenatal period is unique to NLS9. Additionaly, the ichthyotic skin changes are emphasized to be the characteristic manifestation of NLS; further, the limb anomalies are attributed to reduced intrauterine movements due to tight skin, developing as a result of ichthyosis10,11.
The clinical characteristics of the cases reported in the literature and the current case are summarized in Table 14,12-18. In our case, most of the characteristic findings of NLS, namely ichthyosis, generalized edema and limb anomalies, were observed. Additionally, pachygyria, facial dysmorphism, hypoplastic testes and bilateral cryptorchidism were noted. Microcephaly, which is the most commonly (83%) observed central nervous system defect and intrauterine growth retardation observed in 79% of the previous cases, was not noted in our patient. A similar case has not previously been reported in the literature. Except for intrauterine growth retardation and microcephaly, Thakur et al.19 described a case with similar features of our case. However, unlike our case, their case was a stillborn infant. Another case without intrauterine growth retardation shared findings, such as ichthyosis, abnormal facial features, hypoplastic genitalia and bilateral cryptorchidism, with our patient. However, that patient had microcephaly and lived only 5 days after delivery13.
Most infants with NLS are stillborn or die soon after birth, within minutes to a few hours due to chest constriction, infection or neurologic complications18. However, one of the original patients reported by Neu et al.1 survived 7 weeks. Horn et al.6 also reported two siblings with extreme microcephaly, severe growth and mental retardation, flexion contractures, ichthyosis and mild intrauterine growth retardation who lived beyond 10 months of age; hence, they suggested that there may be a milder variant of this syndrome. A more recent report described a patient with characteristic features of NLS who remained alive at 6 months of age without further follow-up findings5. In our patient, none of the above complications were observed. He had no major health problems during the next 3 years of follow-up, except for mild mental/motor retardation, ichthyosis, facial dysmorphism and limb anomalies.
RD is a lethal genodermatosis that results from mutations in the LMNA or ZMPSTE24 (FACE1) genes20. This syndrome shares some phenotypic features to those of our patient, namely, tightly adherent, thin and translucent skin, joint contractures and abnormal faces. We conducted a molecular genetic analysis for related genes in our patient and found no mutations. Lamellar ichthyosis was another condition that we considered in the differential diagnosis. Our patient exhibited phenotypic characteristics, including collodion membrane, eclabium and ectropion, which were similar to lamellar ichthyosis. However, the generalized, grayish-brown, strikingly quadrilateral scale replacing the collodian membrane over the first few months of life, scarring alopecia, heat intolerance and palmoplantar keratoderma, which are the additional features of this condition, were not observed in our case. Therefore, although we could attribute limb anomalies of our patient to ichthyosis, the presence of genital hypoplasia, pachygyria and facial dysmorphic features other than ectropion and eclabium indicates a syndrome or a new entity that is more than a simple lamellar ichthyosis. On the other hand, we should conduct a molecular genetic analysis for transglutaminase-1 gene; however unfortunately, due to technical insufficiency, we were not able to perform the analysis.
History of parental consanguinity and multiple affected siblings in some families suggests an autosomal recessive inheritance pattern in NLS4,14,18. Therefore, not surprisingly, this syndrome is frequently reported from countries with high rates of consanguineous marriages, such as Turkey. Indeed, parental consanguinity was present in all the previous cases reported from Turkey10,12-16. Additionaly, multiple affected siblings were reported in two families12,16. Chromosomal analyses of the reported cases were normal and the precise genetic basis currently remains unknown. Animal models of RD, a genodermatosis having similar phenotypic features with NLS, had mutations on chromosomes 6q and 9p4. It was suggested that mutations on the same chromosomes may be responsible for the pathogenesis of NLS; future human gene discovery should focus on those chromosomes4,18.
In summary, it is clear from the patients reported in the literature that NLS represents a heterogenous phenotype. The present case had nearly all the features of NLS, namely, congenital ichthyosis, facial dysmorphic features, limb anomalies, genital hypoplasia and pachygyria. However, we suggest that long survival and mild mental/motor retardation without intrauterine growth retardation and microcephaly indicate that our case is peculiar and therefore, may represent a mild form of NLS. However, it is still possible that these findings may be the features of a distinct genetic entity. We conclude that newborns with ichthyotic skin changes should be examined and followed for minor clinical anomalies; furthermore, NLS should be included in the differential diagnosis.

 XML Download
XML Download