

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Melanoma, a neoplasm of the skin that arises from the transformation of melanocytes, is the most lethal of cutaneous malignancies1. Many factors are now known to contribute to the formation of melanoma, including exposure to ultra-violet radiation, skin type, hair/eye color, the presence of dysplastic nevi and/or increased nevi number, and family history of melanoma2. Mutations of several oncogenes, including NRAS, BRAF, CKIT and cyclin-dependent kinase (CDK) and tumor suppressors such as CDKN2A (p16Ink4A) and p53, are linked to melanoma development3-5. BRAF mutations are found in 70% of all melanomas and more than 90% of these mutations carry a single missense mutation consisting in an activating substitution of glutamate for valine at position 600 (BRAFV600E) within the kinase domain of this protein6,7. CDKN2A is a gene involved in familial melanoma pathogenesis and a germline mutation is found among melanoma-prone families8. Additionally, rare germline mutations of CDK4 are exclusively found in individuals with genetic predisposition to melanoma9.
Increased survival of melanoma patients has been observed over the recent decades, which may be due to earlier detection (and subsequent surgical resection) of primary melanoma lesions. Yet, therapeutic options for unresectable or metastatic melanoma remain extremely limited. Until recently, dacarbazine and interleukin-2 (IL-2) were the only two agents approved by the US Food and Drug Administration (FDA) for the treatment of metastatic melanoma. In 2011, two additional drugs were approved by the FDA: Vemurafenib (Plexxikon/Zelboraf™; Roche, Basel, Switzerland) and Ipilimumab. Vemurafenib is an oral agent that targets mutant BRAF, while Ipilimumab is a fully human monoclonal antibody against cytotoxic T-lymphocyte antigen 4 (CTLA-4) that is expressed on the surface of activated T cells. This novel immunotherapy increases cell-mediated immunity by blocking the key immune checkpoint molecule CTLA-4, thus enhances natural antitumor immune response. Despite early therapeutic benefit of these drugs, materialized by rapid and spectacular regression of tumors, resistance rapidly occurs, with brutal tumor relapse.
Melanoma displays multifactorial etiology and its genetic and immunological background has not yet been fully elucidated. In vitro studies indicate that cultured melanoma cell lines produce excessive levels of cytokines and growth factors with pleiotropic biological activities10. Among them, Transforming growth factor β (TGF-β) functions as an autocrine and paracrine factor that drives many cellular processes including tumor growth, invasion, escape from immune surveillance, angiogenesis and metastasis11. We herein provide in-depth analysis of the relationship between melanoma progression and TGF-β signaling.
TGF-β SIGNALING
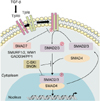
The canonical TGF-β signaling cascade is well characterized. Briefly, TGF-β ligands bind to the type II transmembrane receptor serine/threonine kinases (TβRII), which in turn assembles with, phosphorylates and activates type I receptor (TβRI; ALK5). Activated TβRI phosphorylates the downstream effectors SMAD2 and SMAD3, which then associate with SMAD4 (Fig. 1). This new complex accumulates in the cell nucleus where it regulates the transcription of various target genes. SMAD7, an inhibitory SMAD, interferes with TGF-β signaling by several mechanisms: it can bind TβRI to prevent SMAD2/3 phosphorylation and activation, or recruit the protein phosphatase PP1/GADD34 or the E3 ubiquitin ligases SMURF1 and SMURF2 to the receptor complex, leading to either receptor dephosphorylation or proteasomal degradation, respectively11,12. TGF-β pathway potentially activates other signaling pathways in a non-canonical manner, including the mitogen-activated protein kinases (MAPKs) and PI3K/AKT pathways11.
TGF-β IN MELANOCYTES AND MELANOMA
In normal epithelial tissues, TGF-β acts as a potent cell growth inhibitor. Thus, it is generally accepted that during early stages of carcinogenesis, tumor suppressive effects of TGF-β are mainly driven by the induction of a cytostatic program. TGF-β upregulates cyclin-dependent kinase (CDK) inhibitors (p21, p15, p27Kip1 and p57Kip2), while repressing pro-proliferative factors including c-MYC and inhibitor of differentiation (ID) family members11. In addition to its cytostatic effects, the TGF-β/SMAD cascade also regulates the expression of several pro-apoptotic genes13, which also contribute to its tumor suppressor activities.
In advanced tumors, TGF-β signaling favors cancer cell proliferation and dissemination, peri-tumoral angiogenesis, epithelial to mesenchymal transition (EMT) of tumor cells and tumor escape from immune surveillance, thus acting as tumor promoter.
In normal melanocytes, TGF-β acts as a potent inhibitor of proliferation and differentiation14. TGF-β1 represses the paired-box transcription factor PAX3, which cooperates with the homeobox transcription factor SOX10 to induce melanocyte differentiation15. TGF-β may also interfere with melanocyte maturation by inhibiting of microphtalmia transcription factor (M-MITF), a master transcription factor specific of the melanocytic lineage that controls cell survival, migration and differentiation16.
Cell autonomous activation of the TGF-β pathway in melanoma cell lines has been well documented17,18. While normal melanocytes only express TGF-β1, melanoma cells express and secrete high quantities of the three TGF-β isoforms, TGF-β1, TGF-β2 and TGF-β312. TGF-β ligands are able to induce their own expression through a positive amplification loop. Furthermore, treatment of melanoma cell lines with exogenous TGF-β1 induces the secretion of active TGF-β1 and TGF-β219. Consistently, plasma levels of TGF-β are found elevated in melanoma patients, in parallel with metastatic progression20. Also, TGF-β2 expression was found to correlate with tumor thickness and invasiveness21.
Genome-wide expression analysis of nearly one hundred human melanoma cell lines identified two groups with very distinct gene expression profiles associated with distinct behaviors in vitro: the first group is characterized by high expression of neural crest and melanocytic differentiation markers and exhibits a proliferative, poorly invasive phenotype, while the other group is characterized by the expression of a number of genes whose concomitant expression is reminiscent of a TGF-β signature, and associated with a highly invasive phenotype and low proliferation rate22.
Several studies have described functional SMAD signaling in melanoma. Basal phosphorylation of SMAD3 is detectable in vitro in melanoma cells, demonstrating a constitutive activation of the TGF-β receptors. Furthermore, TGF-β blockade by means of either a neutralizing pan-TGF-β antibody17, SMAD7 overexpression23, or treatment of cells with a TβRI small molecule inhibitor24, inhibited basal SMAD3/SMAD4-dependent transcriptional activity in melanoma cells. Also, exogenous TGF-β was shown to induce further SMAD3/SMAD4-dependent transcription, demonstrating the functionality of the entire TGF-β signaling cascade. In human melanocytic lesions, high levels of nuclear phosphorylated SMAD2 are found in benign melanocytic nevi, melanomas in situ, and primary invasive melanomas, suggesting that tumor cell autonomous hyperactivation of the TGF-β/SMAD pathway can be causally related to melanoma progression in the skin, and may participate to the critical switch from radial to vertical growth during melanoma progression18. Interestingly, it has also been shown that phosphorylation of SMAD2 in melanoma cells is associated with SMAD2 phosphorylation in neighboring keratinocytes, indicating that melanoma-derived TGF-β affects the microenvironment and adjacent cells in a paracrine fashion25.
Intron 6 of the TRPM1 gene (which encodes Melastatin, a calcium permeable cation channel that may play a role in melanin synthesis) hosts the gene for miR-211, whose expression is restricted to the melanocyte lineage26. miR-211 and melastatin share the same promoter and are expressed coordinately in melanocytes and melanomas. TRPM1 is coordinately expressed with miR-211 in melanocytes and melanomas. It is robustly expressed in benign and dysplastic nevi and in melanomas in situ, but its expression is heterogeneous in invasive melanomas and is downregulated in melanoma metastases27,28. Interestingly, it has been shown that reducing miR-211 levels is sufficient to convert a non-invasive melanoma to an invasive melanoma, and that TβRII is one of the direct targets of miR-21129. Yet, TGF-β has been implicated in the metastatic spread of melanoma12, suggesting that increased TGF-β signaling through TβRII expression might enhance melanoma invasion and motility when miR-211 levels are low.
MELANOMA RESPONSIVENESS TO TGF-β
Melanoma resistance to TGF-β-mediated growth suppression
While normal melanocytes are growth-inhibited by TGF-β, melanoma cells exhibit a variable degree of resistance to its antiproliferative activity. The mechanisms underlying evasion from a cytostatic response to TGF-β in these cells remain somewhat elusive. Alterations in the components of the TGF-β signaling in melanoma, including loss of expression or mutation of TβR and SMADs, are very rare12 and therefore do not explain the mechanisms of resistance of melanoma cells to TGF-β-mediated growth arrest and apoptosis.
Various mechanisms have been suggested to explain a possible attenuation or inhibition of the TGF-β signaling cascade in melanoma cells. For example, the type III TGF-β receptor CD105 (also called endoglin) was reported to bind TGF-β1 in melanoma cells and to contribute to the regulation of the anti-proliferative effects of TGF-β30,31. In melanoma cells deficient in filamin, a cytoskeletal actinbinding protein, TGF-β/SMAD signaling is defective, suggesting that filamin, which was identified as a SMAD-interacting protein, may serve as an anchor protein that allows localization of SMAD proteins near cell surface receptors, thus facilitating their phosphorylation and subsequent activation32. More recently, distal-less homeobox (DLX2), identified as a novel TGF-β-inducible transcription factor in B16 melanoma cells, was shown to exert protective function against the growth suppressive effects of TGF-β33. Also, forced expression of H11/HSPB8, an atypical heat shock protein with kinase activity that inhibits the growth of melanoma cells through activation of TGF-β activated kinase 1 (TAK1)-dependent death pathways, is silenced through aberrant promoter methylation in melanoma cells, thus contributing to resistance to TGF-β-mediated cytostasis34.
High levels of the oncoproteins SKI and SNON are detected in melanoma35,36. These proteins have been described as potent inhibitors of TGF-β signaling, acting both in the cell nucleus where they recruit transcriptional co-repressors to active SMAD complexes, and in the cytoplasm where they sequester SMAD proteins11. While it may be easily interpreted that such high expression of TGF-β pathway inhibitors is an obvious mechanism for melanoma cells to escape the growth inhibitory activity of TGF-β, reports from various laboratories indicate a complex regulatory network. In some melanoma cell lines, SKI or SNON knockdown inhibit cell and xenograft tumor growth36-38. Melanoma inhibitory activity (MIA), a protein expressed in melanoma but not in melanocytes39, has been shown to upregulate SKI and SNO expression via the MAPK/ERK cascade. This, in turn, was suggested to disrupt TGF-β signaling in melanoma cells40. It needs to be emphasized, however, that TGF-β signaling is functional in melanoma and that autocrine activation of the pathway in melanoma cell has been well documented12,17,18,41, underlying the complexity of TGF-β signaling in melanoma progression. Regarding SKI and SNON, TGF-β is a potent and rapid inducer of SKI and SNON degradation by the proteasome in a variety of cancer cell lines, including melanoma35,42. In the latter study, active and efficient TGF-β signaling and responsiveness was verified in melanoma cell lines, associated with rapid degradation of SKI and SNON proteins, within minutes after TGF-β stimulation. This event takes place not only in melanoma cells but also in various breast carcinoma cell lines42. Also, SKI knockdown prior to exposure to TGF-β did not significantly affect the extent of TGF-β responsiveness. Together, the latter studies indicate that TGF-β efficiently counteracts the ability of SKI and SNON to inhibit metastasis by inducing their degradation by the proteasome.
A number of other possible mechanisms of resistance to TGF-β may exist in melanoma, yet their functional relevance in disease is still ambiguous given the efficacy of TGF-β inhibitors to antagonize experimental melanoma progression24. For example, phosphorylation of the SMAD2 and SMAD3 linker regions was found to be high in both melanoma cell lines and melanoma samples43. Such event has previously been identified as important in the control of SMAD2/3 activity, leading to downregulation of TGF-β-responsive genes such as p21, p15 and c-MYC, and subsequent escape from TGF-β-induced cell growth arrest19. Yet, activation of other TGF-β target genes, such as PAI1, was not modified, suggesting additional, currently unknown, levels of activity control. The mechanisms responsible for SMAD2/3 linker domain phosphorylation may include MAPKs (p38, JNK and ERK)44-46, as well as GSK3β47 and CDKs48,49. This is compatible with the fact that melanomas are characterized by a high incidence of activating mutations of the BRAF and NRAS oncogenes, which lead to constitutive activation of MEK/ERK signaling, and thus could participate in SMAD2/3 linker phosphorylation. Also, stable overexpression of NRAS in melanoma cells was found to interfere with TGF-β-induced growth inhibition via alteration of Rb protein phosphorylation50.
ID proteins have been shown to enhance cell cycle progression as they interfere with bHLH, ETS and PAX transcription factors (that negatively regulate CDK inhibitors such as p15Ink4b) and bind tumor suppressor proteins like Rb51. Interestingly, ID2 overexpression was shown to protect melanoma cells from TGF-β suppression of proliferation. ID2 is differently expressed in melanoma cells according to their proliferative vs. invasive phenotype. ID2 may be downregulated by TGF-β in proliferative cells that respond to its growth inhibitory effect, while TGF-β cannot repress ID2 expression in invasive cells52. PAX3, which is repressed by TGF-β in primary melanocytes, also participates to melanoma cell survival53. In murine B16 melanoma cells, PAX3 overexpression was found to desensitize cells to TGF-β-induced growth inhibition15, thus representing another potential mechanism of resistance to TGF-β anti-proliferative effects.
Autocrine TGF-β signaling promotes melanoma progression and metastasis
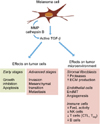
Melanoma cells secrete high levels of TGF-β that acts on tumor cells and their microenvironment by both autocrine and paracrine mechanisms, to initiate or amplify various processes, either cell-autonomous or non cell-autonomous, that contribute to tumor progression (Fig. 2).
In epithelial tumor cells, TGF-β pathway promotes an EMT, a critical multistep process that converts epithelial cells into motile and invasive mesenchymal-like cells. This mechanism is believed to allow cancer cells to escape from the primary tumor and to become metastatic54. During the transition from radial growth phase to vertical growth phase, melanoma cells undergo a process that may be considered a pseudo-EMT whereby melanoma cells acquire a fibroblastoid morphology55, via mechanisms that recapitulate many of the molecular characteristics of EMT56. In this context, TGF-β has been shown to promote melanoma cell pseudo-EMT through up-regulation of matrix metalloproteinase MMP9, β1 and β3 integrin and down-regulation of the epithelial marker E-cadherin.
The role of autocrine TGF-β/SMAD signaling in controlling the invasiveness and metastatic potential of melanoma cells has been described in several studies from our laboratory. In melanoma cells, we showed that overexpression of SMAD7, an inhibitory SMAD that interferes with TGF-β receptor function, decreases their capacity to form colonies in an anchorage-independent manner, reduces MMP2 and MMP9 secretion and Matrigel-invading capacity, and delays xenografted tumor growth in immuno-compromized mice57. Moreover, melanoma cells overexpressing SMAD7 were less prone than their wild-type counterparts to form experimental bone metastases following intraventricular inoculation in mice, highlighting the role of TGF-β signaling in metastatic progression23. Mechanistically, SMAD7 was found to induce β-catenin degradation and promote homotypic interactions with N-cadherin between melanoma cells, and to stabilize their association with neighboring dermal fibroblasts, an additional mechanism that likely attenuates invasion58. Another approach consisted in using the small molecule TβRI inhibitor SD-208 to interfere with TGF-β signaling. This pharmacologic inhibitor efficiently blocked TGF-β-induced SMAD3 phosphorylation and SMAD3/4-specific transcription, as well as Matrigel invasion by melanoma cells. Furthermore, SD-208 inhibited the expression of the TGF-β target genes parathyroid hormone-related peptde (PTHrP), IL-11, connective tissue growth factor (CTGF), and RUNX2, all involved in the development of osteolytic bone metastases. Systemic administration of SD-208 to nude mice prior to melanoma cell inoculation dramatically inhibited the establishment of osteolytic bone metastases. Also, SD-208 treatment significantly reduced the growth of established osteolytic lesions in the same mouse model of bone metastases24. Most recently, we found that halofuginone, an alkaloid that interferes with TGF-β signaling through the induction of SMAD7 and repression of TβRII expression59, inhibits melanoma experimental metastasis to bone and brain60.
Taken together, these results demonstrate that therapeutic targeting of TGF-β may prevent the development of melanoma bone metastases and decrease the progression of established osteolytic lesions.
We also demonstrated that GLI2, a transcription factor involved in the hedgehog (HH) signaling cascade, is a direct transcriptional target of TGF-β/SMAD signaling in various cell lines, including melanoma61,62. Accordingly with numerous studies that have established the pro-oncogenic role of GLI2 in various malignancies, GLI2 is highly expressed in aggressive melanoma cells where it regulates their invasive capacity by upregulating MMP2 and MMP9 expression. In melanoma cells, GLI2 expression is driven, at least in part, by TGF-β, and is associated with a loss of E-cadherin expression63. In addition, GLI2 is a potent inducer of WNT5A and PTHrP expression, two proteins involved in the metastatic process64-66.
TGF-β also induces the expression of integrins, proteases such as cathepsin B and matrix remodeling proteins like SPARC that may be involved in the acquisition of mesenchymal traits that favor metastatic dissemination. TGF-β1 contribution to cathepsin B-mediated melanoma invasiveness is highly Secreted Protein Acidic and Rich in Cystein-dependent67. SPARC also cooperates with interferon (IFN)-γ to repress the expression of a disintegrin and metalloproteinase (ADAM)1568. Yet, ADAM15 may have tumor-suppressive functions in melanoma as it was shown to reduce the formation of lung metastases in mice and to reduce migration and invasive capacity of melanoma cells in vitro69. In another study, genome-wide microarray analyses of invasive human melanomas and benign nevi revealed up-regulation of cathepsins B and L, MMP-1 and MMP-9, and urokinase- and tissue-type plasminogen activators. The mRNA levels of cathepsins B/L and plasminogen activators, but not MMPs, correlated with metastasis. Primary melanomas and benign nevi revealed that cathepsin B is predominantly expressed by melanoma cells while cathepsin L is preferentially expressed by tumor-associated fibroblasts surrounding the invading melanoma cells. Cathepsin B was shown to be important for both the production and secretion of TGF-β signaling, which in turn activates fibroblasts and subsequent invasive capacity of melanoma cells70.
In spindle tumor cells including melanoma cells, TGF-β was shown to induce the expression of fascin, an actin-bundling protein critical for metastasis. By stabilizing the actin cytoskeleton within invadopodia and filopodia, fascin coordinates the invasion and metastasis promoted by TGF-β71.
TGF β induced protein (TGF-βI) is a TGF-βinducible, secreted extracellular matrix (ECM) protein shown to impair the adhesive contacts of melanoma cells with the ECM molecules fibronectin, type I collagen and laminin. TGF-βI was found to localize with the fibrillar fibronectin/tenascin-C/periostin structures that characteristically surround the invasive fronts of melanomas72.
Several cytoplasmic proteins have been identified that interact with the TGF-β receptors. Aberrant expression and/or regulation of these receptor interactors could interfere with TGF-β signaling and promote tumor development. In Xenopus, the pseudoreceptor BAMBI (bone morphogenetic protein and activin membrane-bound inhibitor) is a naturally occurring truncated type I receptor that lacks a kinase domain. It can interact with TGF-β in the presence of TβRII, binding TβRI and TβRII and disrupting ligand-induced receptor heterodimerization73. Interestingly, NMA, a mammalian orthologue of BAMBI, was identified by differential display analysis as decreased in highly metastatic melanoma74, suggesting that derepression of TGF-β signaling following a decrease of NMA expression could account for the increased metastatic potential of these cells.
Paracrine effects of TGF-β on the tumor microenvironment
Paracrine secretion of TGF-β may modulate the tumor microenvironment for the benefit of melanoma growth, invasion and metastasis, especially through its capability to activate stromal fibroblasts, its immune-suppressive effects and pro-angiogenic properties.
TGF-β-activated stromal fibroblasts can stimulate ECM protein production, including collagen, fibronectin, tenascin and α2-integrin, thus providing a scaffold that promotes melanoma cell survival, migration and metastatic progression75. Interestingly, autocrine as well as paracrine TGF-β may also promote endothelial-to-mesenchymal transition (EndMT), a process whereby endothelial cells acquire mesenchymal features and contribute to the accumulation of fibroblasts in the tumor microenvironment that facilitates tumor progression in a B16F10 melanoma model76.
During melanoma progression, tumors acquire a rich vascular network due, at least in part, to paracrine secretion of TGF-β that allows the recruitment and proliferation of endothelial cells for the development of new blood vessels. In melanoma cells, TGF-β-induced angiogenesis involves up-regulation of several pro-angiogenic factors, including IL-8 and vascular endothelial growth factor (VEGF)77.
Periostin is a TGF-β-induced ECM protein involved in cell invasion, metastasis and angiogenesis. In a murine model of human melanoma, the use of an antibody directed against a periostin epitope that corresponds to a binding site for integrins significantly reduced tumor growth and angiogenesis78.
Moreover, treatment of a B16 melanoma model with a chimeric protein derived from the fusion of IL-2 and the soluble extracellular domain of TβRII resulted in reduced angiogenesis and tumor growth79.
TGF-β negatively regulates the activity of immune cells and plays an important role in the control of T-cell functions and proliferation. For example, TGF-β blocks the production of IL-2 (a lymphokine that activates T cells, natural killer (NK) cells and other immune cells) and cell cycle regulators in T cells. TGF-β also controls the expression of effector molecules produced by cytotoxic T lymphocytes, and is an important differentiation factor for regulatory T lymphocytes (Treg), which are present in high quantities in tumors including melanoma and contribute to TGF-β-mediated immunosuppression11.
Melanoma cells, but not normal melanocytes, express monocyte chemoattractant protein-1 (MCP-1), a potent chemokine that recruits macrophages80 that in turn infiltrate melanomas and produce factors that help tumor invasion, angiogenesis and immunosuppression81. IL-10 expression has been shown in vitro in many melanoma cell lines82, and in vivo, in primary and in metastatic melanoma lesions83. Also, elevated levels of IL-10 have been observed in patients with advanced melanoma and correlate with tumor progression84. TGF-β was shown to increase the expression of MCP-1 and IL-10 in melanoma cells, an effect mediated by the crosstalk between the SMAD, PI3K/AKT and BRAF/MAPK signaling pathways85.
TGF-β AND MELANOMA THERAPY
Several TGF-β inhibitors, including small-molecule inhibitors targeting type I and II TGF-β receptor activity, monoclonal antibodies neutralizing TGF-β ligands, antisense oligonucleotides blocking TGF-β ligand production and combined therapies utilizing antisense oligonucleotides together with immune-stimulating vaccines, have been developed for cancer therapy and have demonstrated efficacy in preclinical studies11. At this stage, none of these compounds has been approved for clinical use.
In the case of melanoma treatment, several anti-TGF-β therapies have been studied such as the TGF-β2-targeting antisense molecule AP12009 (also called Trabedersen) that has been assessed in phase I/II clinical trial in patients with advanced pancreatic or colorectal cancer and malignant melanoma. AP12009 is safe and well tolerated, and enrolled melanoma patients had somewhat improved median overall survival86.
Because TGF-β is abundantly secreted by tumor cells or by their microenvironment during disease progression, monoclonal TGF-β neutralizing antibodies and soluble TβRII or TβRIII represent a valuable therapeutic option to trap TGF-β ligands. In preclinical studies, these molecules have shown encouraging results, especially for breast cancer treatment87. GC1008 (fresolimumab), an anti-TGF-β monoclonal neutralizing antibody, is under evaluation in a clinical phase I/II study in patients with advanced melanoma. Also, patients with advanced melanoma achieved stable disease or partial responses even if drug-related side effects were reported such as skin rash/lesions including eruptive non-malignant keratoacanthomas and squamous cell carcinomas88. Small molecule inhibitors that target TGF-β receptor kinases are also studied for cancer treatment. A major drawback of these pharmacologic compounds is their high cardio-toxicity in humans, which has taken most of them out of clinical trials89.
As TGF-β exerts a key role in tumor immune evasion, a novel therapeutic approach was developed, consisting in an autologous tumor cell vaccine (FANG™, Gradalis Inc., Carrolton, TX, USA) that incorporates a plasmid encoding both granulocyte-macrophage colony-stimulating factor (GM-CSF) and a bifunctional short hairpin RNAi (bishRNAi) directed against the proprotein convertase furin responsible for TGF-β activation. This strategy aims at downregulating endogenous immunosuppressive TGF-β1 and β2. In a phase I trial in patients with advanced tumors, FANG™ vaccine did not cause severe adverse events, and elicited an immune response correlated with prolonged survival90,91. It is currently tested in phase II trials for the treatment of colon and ovarian cancer, as well as advanced melanoma.
Despite FDA approval of IL-2 for melanoma treatment several years ago, only 5~6% of patients with metastatic melanoma achieve durable complete remissions when treated with high doses of this lymphokine that induces or expands activation of melanoma-specific T-cell responses. Interestingly, IL-2 treatment combined with nanolipogel delivery of a TGF-β inhibitor was found to significant delay melanoma growth and increase survival in tumor-bearing mice, along with an increase in NK and tumor infiltrating lymphocytes (TIL) activity92.
CONCLUSION
The current literature has brought ample and clear evidence for a pathogenic role of TGF-β signaling in melanoma progression to metastasis. TGF-β not only acts on melanoma cells, but also on their microenvironment including stromal fibroblasts, immune and endothelial cells. Crosstalks between these different cell types are crucial for melanoma establishment, progression and dissemination. The TGF-β signaling cascade has thus attracted much attention as a potential therapeutic target. Several TGF-β-targeting agents (combined or not with other drugs) are currently under evaluation in clinical trials for melanoma treatment and have shown promising results in enrolled patients86,88,92. However, it is fundamental to consider the other signal transduction cascades that are known to play a role in melanoma progression, as TGF-β signaling was shown to crosstalk with some of them in epithelial cancers, including the MEK/ERK pathway.
It would be interesting to test the effects of TGF-β inhibitors combined with the new therapeutic drugs specifically targeting mutant BRAF or MEK. Finally, it is essential to take into account the influence of TGF-β on the tumor microenvironment during melanoma progression. Novel strategies that combine anti-TGF-β therapy and specific targeting of proteins involved in tumor cell-microenvironment interactions might also be worth considering.
 XML Download
XML Download