

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Keratinocytes progress through the suprabasal layers during epidermal differentiation, undergoing complex and tightly regulated biochemical modification leading to cornification and desquamation. The development of DNA chips in combination with in vitro models of human epidermal morphogenesis enhances our ability to identify all genes that are involved in the process of epidermal stratification1.
Epigenetics is the study of potentially heritable phenotypical differences in the absence of variation in the genetic code. That genetic variability is important in the etiology of many diseases is well established. However, environmental factors, including diet and lifestyle, modulate that susceptibility in part through epigenetic changes2. Epigenetic phenomena involve both intracellular and intercellular interactions, leading to alterations in reversible phenomena such as cell signaling and DNA modification3. Epigenetics is important in the pathogenesis of many skin diseases4. For common skin cancers, aberrant methylation of tumor suppressor gene promoters is associated with their transcriptional inactivation5. Hypomethylation is associated with activation of systemic autoimmune diseases, such as systemic lupus erythematosus, and scleroderma6. Epigenetic factors may also play a role in the pathogenesis of psoriasis and other inflammatory skin diseases7. However, there are no reports that identify an epigenetic modulation of gene expression during keratinocyte differentiation. We investigated whether epigenetic modulation is involved in keratinocyte differentiation-specific gene regulation.
MATERIALS AND METHODS
Skin samples and preparation
Normal human skin samples were obtained from circumcisions under the written informed consent of the donors, in accordance with a process approved by the Ethical Committee and Institutional Review Board of Chungnam National University Hospital (cnuh 2011-06-104). Subcutaneous fat was promptly removed and strips of skin were incubated, epidermis side up, for 1 hour at 4℃ in phosphate-buffered saline (PBS) containing 0.5 mg/ml of thermolysin (Sigma, St Louis, MO, USA). The epidermis was dissected free from the dermis using forceps, and rinsed in cold PBS. Epidermal fragments were either immediately frozen for total RNA extraction, or incubated in a 1× trypsin-ethylenediaminetetraacetic acid (EDTA) solution (Invitrogen, Carlsbad, CA, USA) at 4℃ under gentle agitation for 15 minutes. The remaining epidermal fragments were rinsed in cold PBS and incubated in another trypsin-EDTA solution. Fetal calf serum (Invitrogen) was added to the suspended cells (10% final concentration). After centrifugation, the cells were frozen as dry pellets. The procedure was repeated twice, leading to three successive fractions of dissociated cells named the T1, T2, and T3 fractions, and a residual fragment named the T4 fraction.
Morphological analysis of epidermal samples
After each trypsin incubation, an aliquot of epidermal fragments was fixed and embedded in paraffin, and sections (10 µm) were stained with H&E.
Reverse transcription polymerase chain reaction (RT-PCR) analysis
Total RNA was isolated from epidermal fragments using an Easyblue RNA extraction kit (Intron, Daejeon, Korea) according to the manufacturer's recommended protocol. Two µg of total RNAs was reverse transcribed using moloney-murine leukaemia virus reverse transcriptase (ELPIS Biotech, Daejeon, Korea). Aliquots of an RT mixture were subjected to PCR cycles with a specific primer set. All primers used in this study were designed using the Primer 3 input (http://frodo.wi.mit.edu/primer3/) and are as follows: keratin 14 (K14) 5'-CAGTTCACCTCCTCCAGCTC and 5'-TCCTCAGGTCCTCAATGGTC, keratin 10 (K10) 5'-CTACTCTTCCTCCCGCAGTG and 5'-TTGCCATGCTTTTCATACCA, cyclophilin 5'-CTCCTTTGAGCTGTTTGCAG and 5'-CACCACATGCTTGCCATCCA. PCR fragments were electrophoresed on 1.0% agarose gel and subsequently visualized using ethidium bromide staining.
Methylated-CpG island recovery assay-assisted microarray analysis
Genomic DNA was isolated from the T1 and T4 fractions using a DNeasy purification kit (Qiagen, Valencia, CA, USA). We sonicated genomic DNA to produce random fragments ranging in size from 100 to 600 bp. Two Microgram of MBD2bt protein was incubated with 1 µg of sonicated genomic DNA in a binding reaction mixture (10 mM Tris-HCl, pH 7.5, 50 mM NaCl, 1 mM EDTA, 1 mM DTT, 3 mM MgCl2, 0.1% Triton-X100, 5% glycerol and 25 mg/ml of bovine serum albumin) for 4 hours at 4℃ on a rocking platform. The DNA-protein complex was precipitated using preblocked nickel-magnetic beads. After washing the pelleted magnetic beads five times in a washing buffer (binding buffer containing 700 mM NaCl), the methylated DNA-enriched DNA fraction was purified using a Qiaquick PCR purification kit (Qiagen).
Enriched methyl DNA was amplified using a whole genome amplification kit (GenomePlex® Complete Whole Genome Amplification Kit, Sigma) as recommended by the manufacturer. A second round amplification was performed using an aminoally-dUTP mixture (Sigma) and a whole genome amplification kit. The amplified products were labeled by coupling with Cy3 and Cy5-monofunctional dye (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Then, dye-labeled DNA samples were purified and quantified using an ND-1000 spectrophotometer (Nano-Drop Technologies, Inc., Wilmington, DE, USA).
After checking the labeling efficiency, each 2.5 to 5 µg of Cy3-labeled and Cy5-labeled DNA target was mixed, and then resuspended in a 2X hybridization buffer with Cot-1 DNA, an Agilent 10X blocking agent, and de-ionized formamide. The arrays were hybridized at 67℃ for 40 h using an Agilent Hybridization oven (Agilent Technology, Santa Clara, CA, USA).
Data acquisition and analysis
Hybridization images were analyzed using an Agilent DNA Microarray Scanner (Agilent Technology) and data quantification was performed using Agilent Feature Extraction software (Agilent Technology). Preprocessing of raw data and normalization steps were performed using GeneSpring 7.3.1 (Agilent Technology). The individual CpG methylation difference was directly compared based on ratios of signals from control sample DNA.
RESULTS
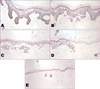
We first applied trypsin to epidermal fragments. Repetitive incubation of human epidermal pieces with trypsin was performed to achieve four suspended cell fractions. Morphological analyses revealed that the T1 fragments were mostly composed of the basal layer, T2 was the spinous layer, and T3 was both the spinous and granular layers. Residual epidermal fragments were mostly composed of the cornified layer (Fig. 1). RT-PCR was used to assess the relative cytokine expression levels in the successive cell fractions. K14 was used as a basal proliferation marker, and K10 as a keratinocyte differentiation marker. Cyclophilin was used to compare total amounts as an internal control. The level of K14 was increased in the T1 fraction, and the level of K10 was increased in the T2, T3, and T4 fractions (Fig. 2). The T1 fraction represented the basal and proliferative layers, while the T2, T3, T4 fractions represented the suprabasal and the differentiation layers.
To investigate epigenetic modulation of gene expression during keratinocyte differentiation, we isolated genomic DNA from the T1 and T4 fractions and applied this DNA to a methylation microarray chip. After hybridization, the microarray slide was scanned and analyzed. Each gene was spotted according to its signal intensity as T1 (Cy3) versus T4 (Cy5). Graphs are shown (Fig. 3) on a log scale using DNA methylation microarray data.
Labeled DNA samples were hybridized to human CpG island microarrays containing 237,000 oligonucleotide probes covering 27,800 CpG islands. To select multiple probes for an enriched genes test, methylation candidate genes were chosen when their probes showed ≥ 2.0-fold in the methylation test, compared with control samples in at least two adjacent probes, allowing a one-probe gap within the CpG islands. We examined the hypermethylation status of these 9 genes: PCDHGA3, FBXL17, SPG20, DUS3L, MRPL36-NDUFS6, PRKD3, PSCD2, C2orf3, FLJ32447 and the hypomethylation status of these 9 genes: ZZEF1, ZNHIT3, ZFAND2A, ZBTB11, YWHAQ, WDR54, VAX1, TXNDC9-EIF5B, and SPCS2. Among these genes, PSCD2 was the most significant gene in hypermethylation field, and YWHAQ was the most reliable gene in hypomethylation field. Detailed information is shown in Table 1.
DISCUSSION
As the keratinocyte differentiation process occurs along a pathway that leads to cell cycle arrest and terminal differentiation, a complex program of gene expression must be coordinated1,8. Many differentiation-related genes, including those encoding transglutaminases 1 and 3, involucrin, cornifin, loricrin, filaggrin, and small proline-rich proteins, are expressed in a temporally regulated manner9,10. New genes such as Brn211, Nkx 2.512, plasminogen activator inhibitor-213 has been suggested to play a role in keratinocyte differentiation.
There are three major molecular mechanisms mediating epigenetic change. They are DNA methylation, Histone modification, and MicroRNA interference4. Methylation of the cytosine and guanine dinucleotides (CpG islands) occurs in the promoter region of approximately 40% of genes in higher eukaryotes. Methylation usually represses gene transcription14, While histone acetylation is generally linked to activation of transcription15. Another mechanism is the effect of nonprotein coding mRNAs on the transcription of other genes and protein synthesis16. The microRNA pathways may be more promising as therapeutic targets, as their effects are more specific than methylation or histone modification.
Knowledge of the contribution of epigenetic mechanisms to the pathogenesis of skin disease has expanded considerably over the last few years, particularly in the field of skin cancer and inflammatory skin diseases9,17. Despite intensive research into skin disease, epigenetic modulation during keratinocyte differentiation is not yet understood.
We used trypsin for epidermal fragmentation, as described previously18 with successive short-term enzyme incubation to progressively detach cells from the deep layers, and to purify the cells. Incubations were performed at 4℃ to stop cellular metabolic activity and to preserve the mRNA pool from degradation. This point is crucial as many growth factors, cell cycle regulators, and transcription factors are encoded by short-lived mRNAs.
We performed methylation DNA microarray analysis with genomic DNA isolated from the basal (T1) and cornified layers (T4). A Methylated-CpG assisted microarray analysis was performed as described previously19. We examined 9 hypermethylated genes and 9 hypomethylated genes. Most of these genes had not previously been associated with keratinocyte differentiation. PSCD2 (pleckstrin homology, Sec7 and coiled-coil domain 2) was the most significant gene in hypermethylation field, and YWHAQ (tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, theta polypeptide) was the most reliable gene in hypomethylation field.
The PSCD2 gene functions to promote activation of adenosin diphosphate-ribosylation factor (ARF) through replacement of GDP with GTP. Members of this family have an identical structural organization that consists of an N-terminal coiled-coil motif, a central Sec7 domain, and a C-terminal pleckstrin homology domain20. The functions of this family include mediating regulation of protein sorting and membrane trafficking20,21. Although there are reports of a regulatory role in development of neuronal processes22, the role of PSCD2 during keratinocyte differentiation has not yet been investigated.
YWHAQ is a gene associated with an adapter protein that is implicated in regulation of a signaling pathway. Binding generally results in modulation of the activity of the binding partner (tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, theta polypeptide)23,24. YWHAQ is directly involved in cellular processes, such as cytokinesis, cell-contact inhibition, anchorage-independent growth, and cell adhesion, processes that often become deregulated in diseases like cancer24,25. Recently, there was a report that YWHAZ, an isoform of YWHAQ, significantly suppressed the growth rate of head and neck squamous cell carcinoma cell lines, and overexpression of YWHAZ in human keratinocytes promotes overgrowth and morphological changes26,27.
We have identified many hypermethylated and hypomethylated genes from differentiated keratinocytes that are involved in epigenetic regulation of keratinocyte differentiation. As this is a preliminary study, more work is necessary to determine whether changes in the methylation status of these candidate genes actually control keratinocyte differentiation. Although extensive work in this field is clearly needed, our preliminary findings highlight the importance of epigenetic modulation in keratinocyte differentiation-specific gene regulation. Furthermore, we provide useful information for future development of novel therapeutic and preventive tools for many skin diseases associated with abnormalities in keratinocyte differentiation.


 XML Download
XML Download