

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Essential thrombocythemia (ET) is a clonal stem cell disease characterized by isolated thrombocytosis and thrombohemorrhagic complications. ET shares phenotypic and pathogenetic similarities with other myeloproliferative disorders (MPD), in particular polycythemia vera and primary myelofibrosis. The clinical presentation of ET is dominated by a predisposition to vascular occlusive events and hemorrhages. Vascular occlusive events include major thrombotic events involving the cerebrovascular, coronary, and peripheral arterial circulation. Deep vein thrombosis also represents a potentially serious and eventually life-threatening event. However, vascular occlusive events can also occur in the micro-vessels where they cause a wide range of clinical symptoms secondary to a transitory suspension of the circulation. They are caused by platelet-mediated transient occlusive thrombosis in the end-arterial circulation1-3. Aspirin-sensitive erythromelalgia is one of the most common microvascular disorders in ET4,5. Livedo reticularis, a characteristic feature of the antiphospholipid syndrome and Sneddon syndrome, has sporadically been observed in ET6,7. We here describe an unusual case of ET primarly presenting with skin symptoms.
CASE REPORT
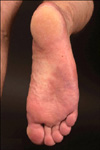
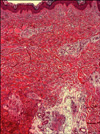
A 40-year-old man presented to our department because of painful skin lesions under both feet. Apart from diatetes mellitus type I and cigarette smoking, the medical history was unremarkable. However, the patient reported increasing B-symptoms such as night sweats and an 11-kg weight loss. On examination, there were violeceous patches on the lateral side of the soles (right>left; Fig. 1) and reticulated purplish-blue patches with broken circular segments on the lower extremities, hips, and lumbar region (right>left; Fig. 2). The lesions on the bottom of the feet were extremely painful in warmth and during walking. Over a 1-year period, the patient had elevated thrombocytes with a mean±standard deviation of 671.166±20.0436/µl (range: 502.000~1,045.000/µl). C-reactive protein and erythrocyte cell count was always normal, but leucocyte counts partly revealed slight leucocytosis. Lactate dehydrogenase was elevated at 343 U/L (normal range, 125~235U/L). Further pathologies included protein S 59% (normal range, 73~143%), factor VIII/C activity 48% (normal range, 50~150%), ristocetin-cofactor 4% (normal range, 50~150%), von Willebrand-factor antigen 26% (blood group non-0: 69~179%), anti-phosphatidylserine IgG and IgM antibodies 51.3 U/ml and 23.3 U/ml, respectively (normally <15 U/ml), and positive smooth muscle autoantibody. Antinuclear antibodies and p- and c-anti-neutrophil cytoplasmic antibodies were within the normal ranges. Polymerase chain reaction analysis of bone morrow revealed a JAK2 gene mutation (codon V617F), and no BCR-ABL transcripts were found. Bone morrow aspirate and histology were consistent with an initial stage of ET. Skin biopsy from the right hip revealed lymphohistiocytic perivascular infiltrates and deep dermal obliterated vessels with fibrinoid necrosis (Fig. 3). Lymph node ultrasound, thoracic and abdomen computed tomography, and cranial magnetic resonance tomography did not reveal remarkable findings. The patient was treated with aspirin (100 mg daily), which led to a immediate dramatic improvement of erythromelalgia. Furthermore, the patient was administered hydroxyurea (1,000 mg daily).
DISCUSSION
The discovery of the JAK2V617F mutation followed by the discovery of JAK2 exon 12, and MPLW515 and TET2 mutations has completely modified the understanding, diagnosis, and management of the classic MPDs. JAK2V617F mutations are present in 90% of patients with polycythaemia vera, 60% of patients with ET, and 50% of patients with myelofibrosis. The diagnostic utility of the MPL and TET2 mutations is limited by their relatively low mutational frequency. There is now strong evidence that these mutations are the oncogenic events that drive these disorders and are responsible for most biologic and clinical abnormalities. The number of JAK2V617F copies (homozygosity vs. heterozygosity) is important in explaining how a single mutation can be associated with several disorders. However, it is still uncertain whether these mutations are sufficient to explain the full development, heterogeneity, and progression of MPDs, or if other genetic or epigenetic events are also necessary1,2.
Patients with ET are at increased risk for arterial and venous thromboembolic events. Arterial ischemic complications occur in about 35% of patients with ET, in particular those with cardiovascular risk factors such as cigarette smoking. Non-atheromatous arterial thrombosis affects large and medium-sized vessels such as cerebral, limb, coronary, and digestive arteries. Moreover, juvenile myocardial infarction or rapid postangioplasty coronary thrombosis may reveal ET. Venous thrombosis is more common in polycytemia vera than in ET. Thromboembolic events occur in about 25~30% of the patients and account for one-third of deaths1-3,8. Decreased levels of protein S, as observed in the present case, may be secondary to chronic clinically occult thrombosis occurring in ET9. Apart from erythromelalgia and livedo reticularis, microvascular circulatory disturbances include Raynaud's phenomenon, acrocyanosis, blue toe syndrome, and cutaneous ulcers or necrotic purpura1,6,7. As also true for the present case, the aforementioned cutaneous signs are frequently the first symptoms of ET and may precede the underlying myeloproliferative disorder by several months. Erythromelalgia is a rare condition of unknown etiology that results in intense, burning pain and redness, primarily of the feet, and, even more rarely, the hands. Most cases are idiopathic (primary erythromelalgia), while others occur secondary to medical conditions such as autoimmune diseases, neurological disorders, and MDS. Symptoms are episodic and can result in severe disability. Triggers, such as exposure to warmth, pressure, or exercise become apparent to those afflicted with this condition. If left untreated, erythromelalgia may progress towards persistent acrocyanosis and even peripheral gangrene4,5.
Another microvascular circulatory condition infrequently associated with ET is livedo reticularis (racemosa-type), which is characterized by a striking violaceous netlike patterning of the skin similar to the familiar livedo reticularis, from which it differs by its location (more generalized and widespread, found not only on the limbs, but also on the trunk and/or buttocks), its shape (irregular, broken, circular segments), and its biopsy results. Histopathologically, only small-to-medium-size arteries of the dermis-subcutis boundary are involved in a stage-specific pattern in livedo reticularis (racemosa-type), particularly showing partial or complete occlusion of the lumen by a plug of lymphohistiocytic cells and fibrin. Livedo racemosa is the typical sign of Sneddon's syndrome and antiphospholipid syndrome7. Interestingly, our patient had livedo reticularis associated with elevated antiphospholipid antibodies highly suggesting an antiphospholipid syndrome. However, patients with ET may show autoimmune epiphenomena, such as elevated antiphospholipid and/or antinuclear antibodies10,11.
Hemorrhagic manifestations in ET are often limited to recurrent skin manifestations, such as subcutaneous hematomas and epistaxis. Hemorrhagic complications are infrequently observed during the course of the condition when appropriate preventive measures are taken. Hemorrhagic symptoms are predominantly observed in patients with very high platelet counts1-3. The hemorrhagic diathesis is not due to impaired platelet function, but rather to an acquired von Willebrand's disease12, which was also found in the present case. In ET, acquired von Willebrand's disease is likely caused by proteolytic reduction of von Willebrand factor multimers. There is an inverse relationship between von Willebrand factor levels and platelet counts. The von Willebrand factor large multimer deficiency appears at platelet counts of 1,000 to 1,500× 109/L, and increases thereafter. Aspirin may unmask a latent bleeding diathesis and may result in severe hemorrhagic complications. It is, therefore, contraindicated in patients with bleeding history and a very high platelet count (in excess of 1,500×109/L leading to the acquisition of von Willebrand's deficiency)1,12. We considered our patient at intermediate thrombohemorrhagic risk, given the presence of diabetes, a platelet count below 1,500× 109/L, and age between 40 and 60 years1,3. Because of this risk profile and acquired von Willebrand's disease, we initiated cytoreduction using hydroxyurea.
In conclusion, the present case highlights the complex nature and diagnostic challenge of MDSs, such as ET, which can involve multiple organ systems and often shows a variety of microvascular complications, coagulation anomalies, and autoimmune phenomena.

 XML Download
XML Download