

 PDF
PDF ePub
ePub Citation
Citation Print
Print


THE SKIN: AN INNATE IMMUNE ORGAN
Our skin is the first barrier against the outside environment. In order to provide an efficient defense the skin is equipped with various innate mechanisms against invading microbial pathogens. Most do not require the specific recognition of the invading pathogen and within cutaneous innate immunity three distinct barriers can be identified: a chemical, a physical and a cellular barrier. While a low pH and small cationic peptides with antimicrobial activity contribute to the chemical shield on the surface of the skin, the stratum corneum forms the initial physical barrier. Epidermal keratinocytes form the first cellular barrier against infectious agents1. These cells together with professional antigen presenting cells such as dendritic cells and dermal macrophages form the first line of cellular innate immunity in the skin. All these cells are equipped with sensors and communicate with each other upon microbial challenge or a danger signal. Subsequently, further immune or pro-inflammatory cascades are triggered providing an adequate and coordinated immune response.
ANTIMICROBIAL PEPTIDES (AMPs): ESSENTIAL PLAYERS IN CUTANEOUS INNATE IMMUNITY
Among the soluble factors secreted onto the cutaneous surface AMPs play a particularly important role in innate immunity. AMPs are evolutionary well-conserved gene-encoded, short (<100 amino acids), amphipathic molecules with hydrophobic and cationic amino acids arranged spatially2. AMPs form predominantly two different secondary structures, disulfide-rich peptides form beta-sheets while linear peptides form alpha-helices3. Their amphipathic structure allows those peptides to be soluble in aqueous environments but also to interact with lipid membranes3. Initially, AMPs were identified as endogenous antibiotics due to their potential to kill various pathogens by disrupting their membranes. They have broad spectrum antimicrobial activity and are able to kill gram-positive and gram-negative bacteria, viruses and fungi4. Keratinocytes and other resident cells in the skin such as eccrine gland cells, mast cells and sebocytes produce and secrete AMPs. In addition, invading immune cells (e.g. neutrophils, natural killer cells) contribute to the pool of AMPs in the skin4-8.
Resident and infiltrating cells synthesize an impressive array of AMPs and to date several hundred of different peptides with antimicrobial function have been identified in human skin9-11. Interestingly, many AMPs were first characterized for other biological activities and their antimicrobial function was later identified. As an example the leukocyte protease inhibitor - also known as α-melanocyte stimulating hormone - exerts antimicrobial activity when tested in culture9.
Importantly, the production of AMPs in the skin is a dynamic defense mechanism: While some AMPs are expressed constitutively in the skin, the production of others is highly increased in danger situations such as skin injury or infections12-15. Furthermore, the expression and function of AMPs are regulated on the transcriptional and post-transcriptional level. Most AMPs are synthesized as pro-peptides and activated after proteolytic cleavage from their precursor molecules16,17. Consequently, induction of transcription and increased processing are needed to provide the skin with enhanced AMP activity.
Two important and well-studied AMP families in human skin are the defensins and the cathelicidins18,19. β-defensin (HBD) 2 was the first skin-derived AMP characterized in man. HBD2 is most effective against gram-negative bacteria whereas HBD3 from the same AMP family has a broader spectrum of antimicrobial action13,20. HBD2 is induced in skin inflammation and infection21 and Gläser et al.22 recently showed that HBD2 and 3 are inducible by ultraviolet-B (UVB) irradiation as well23. In contrast, HBD1 is constitutively expressed in human skin.
CATHELICIDIN LL-37: AN IMPORTANT AMP IN HUMAN SKIN
While there are several gene-encoded defensins, e.g. α- and β-defensins, to date only one single cathelicidin gene has been identified in man and designated cathelicidin antimicrobial peptide (CAMP)24,25. Like many AMP genes CAMP encodes for a pro-peptide which is composed of an N-terminal cathelin domain and a C-terminal peptide with antimicrobial activity26. The first active cathelicidin identified was LL-37 - a 37 amino acid long peptide with broad antimicrobial activity. LL-37 forms an α-helix in aqueous solution which enables the peptide to disrupt both bacterial membranes and viral envelopes9. Even anti-fungal activity of LL-37 in Candida infections was reported27-29. On the skin surface, active cathelicidin peptides such as LL-37 are cleaved from the inactive precursor by serine proteases of the kallikrein family16,19,30.
As LL-37 gains its antimicrobial activity after cleavage from the pro-form processing of cathelicidin by skin proteases is an important regulatory mechanism of cathelicidin mediated antimicrobial activity. In skin, LL-37 is synthesized by epithelial cells but also provided by infiltrating immune cells such as neutrophils which transport LL-37 to infected or wounded skin. In healthy skin, cathelicidin expression is barely detected in keratinocytes. In contrast, during infection or injury cathelicidin production is strongly induced in these cells14.
Apart from the antimicrobial activity LL-37 has additional functions in the activation and the control of immune responses: LL-37 increases cytokine and chemokine liberation from local cells and leucocytes and has chemotactic effects on a large number of immune cells4. In addition, chemokine and cytokine release is induced by LL-37 in mast cells or keratinocytes31. In cooperation with the cytokines LL-37 enhances innate immune responses by multiple pathways4,32. Furthermore, LL-37 enhances the proliferation of endothelial cells and influences angiogenesis33. These attributes complement the antimicrobial functions of LL-37 and have lead to the perception of LL-37 as not only an antimicrobial but an "alarmin"-peptide34.
On a molecular level LL-37 mediates its "alarmin" functions on immune or resident cells in a ligand-receptor mediated or a receptor-independent manner resulting in increased host responses35. In doing so, LL-37 influences adenosine triphosphate-receptor P2X7 and Toll-like receptor (TLR) signaling in immune cells, epidermal growth factor receptor transactivation or intracellular Ca2+ mobilization31,36-38.
The dual role of cathelicidin - the antimicrobial and the alarmin function - suggests a central role for this peptide in cutaneous innate immunity. Consequently, dysfunction of the "alarmin"-function of cathelicidin LL-37 could play a role of in the pathogenesis of inflammatory skin disease to the same extent as impaired antimicrobial activity.
DYSFUNCTION OF CATHELICIDIN AS A CAUSE OF INFLAMMATORY SKIN DISEASE
Emerging evidence suggests that indeed a number of inflammatory skin diseases are characterized by dysregulated expression or function of cathelicidin peptides. In this review, the current knowledge on the role of cathelicidin in the pathogenesis of atopic dermatitis (AD), rosacea, psoriasis and hidradenitis suppurativa (HS) will be presented and discussed.
ATOPIC DERMATITIS
AD is a very common inflammatory skin disease with a chronic course. Patients suffering from AD show an increased susceptibility to infections by viruses, bacteria or fungi and have an altered skin microflora39. A primary defense barrier defect caused by structural defects (e.g. mutations in the filaggrin gene) or by an immunoglobulin E (IgE)-mediated immunonologic disorder with IgE-mediated allergic sensitization and an epithelial-barrier dysfunction as a consequence of the local inflammation are discussed39.
A deficient innate antimicrobial barrier in AD patients was first proposed when an impaired expression of AMPs such as cathelicidin and defensins was detected in lesional skin in AD40. In particular, induction of cathelicidin mRNA transcription in response to wounding is suppressed in AD lesions as compared to healthy skin41. This could be explained by the altered tissue microenvironment in AD skin: Th2 cytokines such as interleukin (IL)-4 and IL-, which are highly elevated in AD skin, suppress cathelicidin induction in keratinocytes42. As itching is a hallmark of AD and scratching results in skin wounding failure to upregulate cathelicidin in response to injury could decrease the cutaneous antimicrobial activity in AD skin41.
Furthermore, it has been hypothesized that the increased rate of microbial superinfections in AD may be caused by reduced AMP expression as a consequence of immunosuppressive therapy. In this context, topically applied corticosteroids and the calcineurin inhibitor pimecrolimus reduce the expression of several AMPs in skin in AD compared with healthy controls43.
In contrast, other groups found enhanced expression of cathelicidin LL-37 mRNA and protein expression in lesional skin compared with non-lesional skin in AD44. Also, non-lesional skin of atopic and non-atopic children shows no significant difference in cathelicidin LL-37 and HBD-2 expression45. Also, the serum levels of cathelicidin LL-37 in AD children did not differ from healthy controls and increased in subgroups with more severe disease46. Finally, serum LL-37 did not differ between patients with or without relevant bacterial superinfection.
Nevertheless, two clinical studies identified subgroups of AD patients with severe infectious complications and a history of dermatitis herpeticatum in the past, which showed defective upregulation of AMPs47,48.
Hence, the role of cathelicidin LL-37 in the pathogenesis of AD is still unclear. Further studies - in particular in subgroups of AD patients suffering from severe AD with infectious complications - are needed to exactly characterize the role of cathelicidin LL-37 in AD.
ROSACEA
Rosacea is another inflammatory skin disease mainly affecting the central portions of the face. The disease often affects fair-skinned individuals and shows a chronic relapsing course. Rosacea occurs mainly in adults around the age of 30 years and typically predominates in females49. The clinical presentations of rosacea include erythema and telangiectasias, pustules and erythematous papules, rarely nodules or edema. The pathophysiology of rosacea is incompletely understood but involves a complex interaction of different factors and pathways leading to a chronic inflammatory and vascular response.
As cathelicidin LL-37 has pro-inflammatory "alarmin" functions and affects vascular growth the expression of cathelicidin was investigated in rosacea recently. Indeed, cathelicidin is strongly increased lesional skin in rosacea compared to the skin of non-affected individuals50.
As mentioned above, processing of the precursor molecule is a crucial step in activating the different functions of cathelicidin peptides. In the skin cathelicidin is processed by serine proteases of the kallikrein family (particularly kallikrein 5 [stratum corneum tryptic enzyme] and kallikrein 7 [stratum corneum chymotryptic enzyme])16. The main resulting peptide is LL-37 - however, LL-37 can be processed further to smaller peptide fragments. Also these smaller peptide fragments exert immune functions but differ in their antimicrobial and immune activating capacities51.
In lesional skin in rosacea the cutaneous protease activity is enhanced and increased expression of serine proteases such as kallikrein 5 can be observed50. Furthermore, variant cathelicidin peptides and smaller fragments can be detected. Thus, in rosacea increased levels of the vasoactive and inflammatory host-defense peptide LL-37 and its proteolytic peptide fragments are found which can be explained by an abnormal cathelicidin production and pathologic protease activity50,52.
Confirming a pathogenic role of cathelicidin in rosacea, injection of these peptide fragments found in skin of rosacea patients into the skin of mice leads to a rosacea-like disease50. In contrast, the isolated increase of protease activity in cathelicidin knock-out mice does not cause dermal inflammation50.
However, the mechanisms underlying the increased cathelicidin production and the enhanced protease activity in skin of rosacea patients are to date unknown. Both seem to be regulated by different signaling pathways with retinoid-, vitamin D- and cytokine-activated cascades playing important roles53.
Cathelicidin LL-37 expression in human keratinocytes is regulated by the vitamin D pathway54. This could explain why rosacea occurs mainly in the face as exposition to ultraviolet (UV) light triggers activation of vitamin D in keratinocytes and subsequent cathelicidin expression54,55.
Recently, a second - vitamin D independent - pathway triggering the induction of cathelicidin synthesis in keratinocytes was identified: In keratinocytes, cathelicidin expression increases upon several external stimuli (such as infection, injuries, UV irradiation, and permeability barrier disruption) which also trigger endoplasmic reticulum (ER) stress. Indeed, ER stress increases CAMP expression via nuclear factor-κB-carbohydrateresponsive element binding proteinα activation independent of vitamin D receptor (VDR) activation demonstrating a novel role for ER stress in stimulating innate immunity56. This again could explain why rosacea patients often report on unspecific triggers (e.g. heat) which would mediate their pro-inflammatory activities through ER stress and cathelicidin induction.
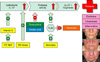
But why is the cutaneous protease activity elevated in lesional skin in rosacea? As mentioned earlier resident cells in the skin such as keratinocytes express receptors sensing pathogen associated molecular patterns (PAMPs). TLRs are one family of PAMP sensors triggering further pro-inflammatory pathways. As an example, ligands for TLR2 are microbial structure molecules such as cell wall fragments but also chitin57,58. In involved skin in rosacea patients keratinocytes express elevated level of TLR257. Furthermore, TLR2 activation in keratinocytes leads to a higher expression of kallikrein proteases and higher protease activity (Fig. 1)57.
And what is the trigger for TLR2 in rosacea? Demodex mites are frequently found on the skin of rosacea patients and skin inflammation correlates with mite density59. Possibly, chitin released from these mites could serve as the trigger for TLR2 on keratinocytes and link demodex with increased protease activity and cathelicidin induced inflammation in rosacea (Fig. 1).
Together these observations possibly identify a complex pro-inflammatory cascade involving demodex, TLRs, cathelicidin and proteases in the pathogenesis of rosacea. Nevertheless, several of these mechanisms could be exploited for novel therapies to be discussed in the following paragraphs.
PSORIASIS
Psoriasis is a third inflammatory skin disease associated with abnormal expression and activity of AMPs60. An autoinflammatory reaction is suspected to play a major role in the course of the disease however the triggers of inflammation in psoriasis remain unknown.
Cathelicidin LL-37 is strongly increased in skin in psoriatic plaques and recently LL-37 was identified as a critical factor for the activation of an auto-inflammatory cascade in psoriasis61. LL-37 isolated from lesional psoriatic skin forms complexes with human self-DNA. These LL-37/self-DNA complexes are sensed by dermal plasmacytoid dendritic cells (pDCs) through TLR961. In turn, activated pDCs secrete large amounts of interferon α to activate further T-cell mediated immune responses. Thus, LL-37 converts inert self-DNA into a potent trigger of interferon production by pDCs in psoriatic skin61.
Subsequently, the capacity of LL-37 to bind to self-DNA and influence inflammation prompted further studies in psoriasis patients. DNA is not only sensed by TLRs but also by absent in melanoma 2 (AIM2), a cytosolic DNA receptor which upon activation triggers inflammasome activation and IL-1β secretion62. AIM2 expression and IL-1β secretion is high in psoriatic lesions but not in healthy skin62. Furthermore, abundant cytosolic DNA was detected in keratinocytes in psoriatic lesions, which triggered the release of IL-1β via the AIM2 inflammasome62. Again, LL-37 bound cytosolic DNA but this time neutralized its pro-inflammatory activity: Cytosolic DNA complexed with cathelicidin LL-37 did not activate the AIM2 inflammasome and subsequent IL-1β secretion was inhibited.
Thus, these studies revealed a contrasting role of cathelicidin in self-DNA mediated inflammation in lesional skin in psoriasis: While dermal LL-37 binds self-DNA and mediates activation of pDCs, epidermal LL-37 complexes with cytosolic DNA in keratinocytes and blocks inflammasome activation and IL-1β release. To date it is unclear which effect - the pro- or the anti-inflammatory - of cathelicidin LL-37 is predominating. Studies in patients under treatment could contribute to the understanding of the role of LL-37 in psoriasis. These studies will be discussed in more detail in the following paragraphs.
HIDRADENITIS SUPPURATIVA
HS is a chronic inflammatory skin disease often resulting in exzessive scarring and fistulation. The axillar, gluteal, inguinal and genital skin areas are often affected. As the pathophysiology of HS is unclear, medical treatment is difficult or even frustrating. To date, surgical excision of affected skin areas is the treatment of choice. AMPs are strongly expression and secreted by the apocrine sweat glands, distal hair follicle epithelium, and epidermis in HS63. In particular, immunoreactivity of the cathelicidin LL-37 was increased in HS lesions. The pro-inflammatory functions of LL-37 could trigger local disease excacerbation and thus promote HS development. However, the triggers and pathways of AMP regulated innate immunity in HS are for the most part unclear and require further research work.
CATHELICIDIN AS A NOVEL TARGET FOR THERAPEUTIC APPROACHES IN INFLAMMATORY SKIN DISEASES
The increasing data on the role of cathelicidin and other AMPs in inflammatory skin diseases has prompted the idea that through targeting AMP expression cutaneous inflammation might be ameliorated. In order to influence AMP expression and function the detailed knowledge of the mechanisms and pathways regulation AMP expression in skin is a prerequisite. As mentioned earlier some regulatory pathways have been identified during the past years: As an example, vitamin D directly activates cathelicidin gene CAMP transcription and LL-37 peptide expression in several cell types such as keratinocytes and monocytes64,65. Importantly, increased cathelicidin expression is paralleled by increased cathelicidin peptide activity34. Restoring antimicrobial activity or balancing "alarming" activities of AMPs could be novel goals of topical or systemic treatments for inflammatory skin diseases66.
ATOPIC DERMATITIS
The rate of atopy and in particular the prevalence of AD is very high in industrialized countries. As patients with AD show structural defects in their cutaneous barrier and dysfunction of cutaneous innate immunity leading to microbial superinfections restoration of the antimicrobial defense shield could be beneficial. Vitamin D directly regulates cathelicidin AMP expression in keratinocytes and a link between systemic vitamin D serum levels, sun exposure and atopy has been discussed67.
Indeed, oral supplementation of vitamin D induces cathelicidin production in skin in AD patients68. Also, systemic treatment with a VDR agonist leads to healing of experimental allergen-triggered eczema possibly through effects on the cutaneous barrier69. Topical vitamin D application on human skin also increases cathelicidin immunoreactivity14. Still, to date it is unclear whether AD patients treated with vitamin D show a decreased rate of infectious complications as a consequence of increased cathelicidin expression.
As mentioned earlier, human skin is able to synthesize active vitamin D from precursor molecules under the influence of UV irradiation. Even in a culture dish UV-B irradiation of human keratinocytes supplemented with 7-dehydrocholesterol triggers the synthesis of hormonally active calcitriol, which then differentially affects expression of AMPs cathelicidin and HBD255. In a clinical setting, UVB phototherapy increases the endogenous production of vitamin D in AD patients which is accompanied by healing of eczema lesions70. These effects might be mediated by improved vitamin D balance, the local cytokine network and/or AMP expression70. However, larger, prospective, multi-center studies are needed to clarify the role AMP regulated barrier functions in AD.
ROSACEA
The pathways involved in increased and dysfunctional cathelicidin in the skin of patients with rosacea are complex. However, due to the multiple regulatory steps involved several strategies are possible for topical and systemic therapies (Fig. 1).
As an example, chitin released from demodex mites possibly triggers TLR2 activation and subsequent protease activity in the skin of rosacea patients. Thus, decreasing the load of demodex mites on the skin or blocking of TLR2 could decrease protease activity and cutaneous inflammation. Indeed, inhibition of demodex has been suggested in the treatment of rosacea before and decreased mite density associates with reduced inflammation in clinical studies71. Retinoids, which are commonly used in the treatment of rosacea, block TLR2 activity and could mediate their effects through this mechanism as well (Fig. 1).
The increased protease activity is central to the pro-inflammatory effects of dysfunctional cathelicidin peptides in rosacea50. Hence, topical or systemic therapies inhibiting pathologic protease activity could exert anti-inflammatory effects in rosacea. Indeed, some established and clinically effective therapies probably exert their beneficial effect through this mechanism. As an example, tetracyclines, such as doxycycline, can inhibit proteases and ameliorate cutaneous inflammation in rosacea34,72,73.
Importantly, these anti-inflammatory effects are independent of the antimicrobial effect of e.g. doxycycline and the doses needed for clinical effect are probably lower than those needed to treat infections (Fig. 1).
Azelaic acid - another topical drug commonly used for rosacea74 - might reduce the protease activity in keratinocytes, too75.
Another way to interfere with the outlined pro-inflammatory cascade could be the blockade of increased cathelicidin production in lesional skin in rosacea. As mentioned earlier, vitamin D controls the expression of cathelicidin and UVB triggers activation of vitamin D from precursor molecules65. Thus, the advice to rosacea patients to avoid exposure to sun light might find its scientific basis in those observations.
Also, alterations VDR gene have been described in patients with severe rosacea (rosacea conglobata) suggesting that vitamin D signaling is indeed affecting the pathogenesis of rosacea76. Drugs which interfere with vitamin D signaling and therefore are able to control the expression and processing of cathelicidins might offer a new approach in the management of rosacea77.
PSORIASIS
As cathelicidin might be a central factor in the pathogenesis of psoriasis therapies targeting cathelicidin expression might influence the course of the disease. On the one hand, the blockage of dermal LL-37 or a decrease in dermal cathelicidin deposition might interrupt the vicious circle of cutaneous inflammation induced by elevated levels of LL-37/self-DNA-complexes which leads to pDC activation61,78. On the other hand, increased epidermal cathelicidin could lead to sustained AIM2 inflammasome inhibition and a decrease in cutaneous inflammasome activity.
Expression of cathelicidin in keratinocytes is regulated by vitamin D. Moreover, vitamin D analogs have been used in the treatment of psoriasis for a long time. However, the molecular mechanisms behind their clinical effects were not completely lucid. vitamin D analogs bind to the VDR followed by binding of the VDR binds to vitamin D responsive element in the promoter region of the cathelicidin gene34,66.
Topical treatment with vitamin D analogs, such as calcipotriol, decrease inflammation and reduce morphological changes in psoriatic lesions79. At the same time calcipotriol treatment decreases pro-inflammatory cytokines but strongly increases the expression of cathelicidin66.
Similar to AD, UVB phototherapy is also used for the treatment of affected skin in psoriasis vulgaris. Repeated treatments of psoriasis patients with narrowband UVB significantly decreased skin inflammation in psoriasis patients but at the same time strongly increased vitamin D serum levels and cutaneous cathelicidin expression70. Thus, established therapies targeting the vitamin D pathway reduce inflammation while increasing epidermal cathelicidin expression in psoriatic lesions. The anti-inflammatory effect of cathelicidin LL-37 on the AIM2 inflammasome pathway could be responsible for this effect.
Vitamin D serum levels can also be increased through oral supplementation and some studies have started to investigate if the vitamin D serum levels in patients with psoriasis correlate with cutaneous AMP expression. One study revealed that cathelicidin expression in lesional skin was higher in serum vitamin D sufficient groups compared to serum vitamin D deficient groups80. Apparently, vitamin D serum levels and cathelicidin expression were not sufficient to inhibit inflammation. Therefore, more prospective interventional studies testing, whether increasing vitamin D serum levels affects the course of psoriasis are needed.
CONCLUSIONS AND OUTLOOK
Many inflammatory skin diseases such as AD, psoriasis and rosacea are characterized by dysregulated synthesis of AMPs. Cathelicidin LL-37 is one important AMP found in skin. As cathelicidin exerts antimicrobial but also immune functions dysregulated expression and processing of this AMP is involved in the pathogenesis of chronic inflammatory skin diseases. As the regulatory mechanisms of cathelicidin gene regulation and peptide processing become clearer strategies to influence these processes emerge. Furthermore, established therapies such as topical and systemic treatments for rosacea or psoriasis might mediate their effects through their impact on cathelicidin. Further research could identify novel targets and mechanisms, which could lead to innovative treatments for inflammatory skin diseases, through their effects on cathelicidin.
 XML Download
XML Download