

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Vitiligo is an acquired depigmentary disorder of the skin resulting from the loss of functioning epidermal melanocytes. The etiology involves multiple pathogenetic factors, including neural theory, impaired antioxidative defenses, mitochondrial membrane lipid peroxidation, a link and temporal sequence between oxidative stress and autoimmunity, and a genetic predisposition, most of them acting in concert1,2.
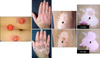
Most studies on vitiligo have concentrated on the abnormality of melanocytes rather than the abnormality of keratinocytes, although epidermal melanocytes form a functional and structural unit with neighboring keratinocytes. Cell-to cell interactions play an important role in homeostasis and regeneration of adult tissues. Cross-talk between has been observed between keratinocytes and melanocytes. In fact, direct cell-to-cell contact stimulates in vitro proliferation and differentiation of melanocytes3. Growth factors produced by adjacent keratinocytes regulate the proliferation and differentiation of melanocytes3. Therefore, damage to keratinocytes might have a significant effect on melanocyte survival. Autologous epidermal grafting is a popular surgical method to replace melanocytes and treat stable vitiligo. Although a similar number of melanocytes is transferred to depigmented epidermis, the outcome of transferred melanocytes would be different; melanocytes may survive by proliferation resulting in homogenous pigmentation, may survive without producing homogenous pigmentation, or may survive temporarily and then die (Fig. 1). In addition, complete homogenous pigmentation is usually restored at the donor sites. These results suggest that local factors participate in the survival and/or growth of melanocytes. Because depigmented epidermis contains only a few 3,4-dihydroxyphenylalanine-positive melanocytes or none at all, resident keratinocytes may be the main source of local factors. Although structural abnormalities in keratinocytes are not remarkable in hematoxylin and eosin (H & E)-stained epidermal specimens in patients with vitiligo, structural changes and their effect on vitiligo development are presented in this study.
APOPTOSIS OF VITILIGINOUS KERATINOCYTES
A loss or a decrease of pigmentation is the main clinical finding in patients with vitiligo. No remarkable microscopic changes, except decreased or no melanocytes, are observed on H & E staining. Nonetheless, an electron microscopic examination showed that basal and parabasal keratinocytes degenerate, not only in depigmented but also in normally pigmented skin4,5. The fine structural changes of degeneration seemed to be consistent with either early signs of cellular necrosis or apoptosis. Additionally, anti-keratinocyte antibodies, which have been detected in the sera of patients with vitiligo, result from keratinocyte death during the disease process6. We also previously examined cytokeratin expression using paired depigmented and normally pigmented epidermis obtained from suction blisters of patients with vitiligo. Western blotting showed more numerous lower molecular weight keratin bands, which are not detected in cultured normal keratinocytes either a high or lower calcium concentration, in depigmented compared to normally pigmented epidermal specimens (data not shown). Although it is unclear how these lower molecular weight bands developed, increased keratin proteolysis7 and limited capacity for in vitro polymerization8 have been suggested. In fact, abnormal cytokeratin expression profiles displaying an increase in lower molecular weight polypeptides have been reported for psoriasis9. Based on these results, we examined and compared the differences in keratinocytes between depigmented and normally pigmented epidermis, particularly focusing on keratinocyte apoptosis.
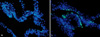
Apoptosis is a distinct mode of cell death, which differs from necrosis in morphology and mechanism, and plays a crucial role in homeostasis. Apoptosis is characterized by cell shrinkage, chromatin condensation, and systemic DNA cleavage and is triggered by various physiological stimuli such as Fas/tumor necrosis factor (TNF) receptors and the loss of survival stimuli10. As apoptotic cells are rapidly engulfed by phagocytes, thereby preventing an inflammatory reaction to the degenerative cell contents11, specific methods such as cell morphology, DNA degradation analysis, DNA end labeling techniques, flow cytometric analyses, and nuclease assays have been developed to detect them. Externalization of phosphatidylserine (PS) and phosphatidylethanolamine is a hallmark of the changes in the cell surface during apoptosis12,13. Permeability of the plasma membrane is also a central difference between necrosis and apoptosis. Based on these results, Annexin V binding as a marker of PS externalization14, large molecular weight DNA binding dyes, such as propidium iodide, and smaller dyes, such as 4-6-diamidino-2-phenylindole, Hoechst 33342 or 33258, have been used singly or in combination to detect apoptosis15,16. However, these assays are only applicable to study isolated cells but not intact tissues. In contrast, methods for detecting the 3'- OH ends of single-stranded DNA breaks, such as terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick end-labeling of DNA fragments (TUNEL) methods, are directly applicable to intact tissue sections. In fact, TUNEL is a method to simplify the identification of apoptotic nuclei in routinely processed tissue sections17. Therefore, we performed TUNEL staining using paired depigmented and normally pigmented skin specimens of patients with vitiligo. The staining revealed that the number of TUNEL- positive cells increased significantly in the basal layers of depigmented compared to normally pigmented epidermis, which was taken from either suction blisters (Fig. 2) or skin biopsies18,19. Because most TUNEL-positive cells in the epidermis did not stain for c-kit, a marker of melanocytes, the TUNEL-positive cells were considered to have originated from keratinocytes. However, TUNEL positivity does not always mean apoptosis. The TUNEL method might detect cells that have not shown DNA fragmentation specific for apoptosis only, but identifies nuclei in areas of necrosis17.
Apoptosis is the best characterized form of programmed cell death. Two major pathways are involved in the initiation of apoptosis in mammalian systems, the extrinsic death receptor pathway and the intrinsic mitochondrial pathway. Caspases are central effectors in the apoptotic signaling pathways, which include the initiation, mediation, execution, and regulation of apoptosis. Caspases are a family of mammalian proteases that mediate many of the morphological and biochemical features of apoptosis. The caspases are synthesized as inactive zymogens whose activation requires limited proteolysis or binding to co-factors20. The extrinsic pathway is initiated by recruitment and activation of caspase-8 in the death-inducing signaling complex (DISC) followed by direct cleavage of downstream effector caspases. In contrast, release of a number of pro-apoptotic factors from the intermembrane space of mitochondria is preceded by the activation of caspase-9 in the intrinsic pathway21. Both the extrinsic and the intrinsic apoptosis signaling pathways converge into a common pathway resulting in the activation of the executioner caspase cascade including caspase-3. The TUNEL-positive cells in skin specimens of patients with vitiligo were stained with anti-active caspase-3 antibody. A Western blot analysis showed that active forms of caspase-9, caspase-8, and caspase-3 were higher in depigmented epidermis when compared to normally pigmented epidermis in patients with vitiligo18. Although active caspase-3 has been detected in the transition zone between the granular and cornified layers of normal human epidermis22, it has not been detected in the basal layers of the epidermis where the TUNEL-positive apoptotic cells were found. Therefore, the TUNEL-positive implies an apoptotic process.
Regulation of apoptosis is an important part of the apoptotic signaling pathways. Pro-apoptotic and anti-apoptotic members of the Bcl-2 family proteins are involved in the regulation. Bcl-2 and Bcl-xL preserve mitochondrial integrity and prevent release of pro-apoptic proteins such as cytochrome c into the cytosol, whereas Bax and Bak promote release of cytochrome c. TUNEL-positive cells in epidermis from patients with vitiligo were also stained with anti-Bax antibody. The levels of Bax expression are higher, whereas those of Bcl-2 are lower in the depigmented compared to normally pigmented epidermis18. Because of the higher levels of Bcl-2 expression in melanocytes and lower levels in keratinocytes23, the lower levels of Bcl-2 in the depigmented epidermis may result from the loss of melanocytes18. The Bcl-2 family member Bid, which connects between the extrinsic and intrinsic pathways after cleavage by caspase-8, is a target of the p53 tumor suppressor24. Whenever p53 is triggered, cell growth is arrested or apoptosis is induced. However, X-linked inhibitor of apoptosis protein and FLICE (fasassociated death domain protein-like interleukin [IL]-1-beta-converting enzyme)-like inhibitory proteins (FLIPs) block the activation of caspases, particularly those involved in the extrinsic pathway25. Cellular FLIP is homologous to caspase-8 and is also recruited to the DISC, resulting in interference with caspase-8 activation at the DISC26. As keratinocytes express higher levels of the anti-apoptotic protein FLIP and the pro-apoptotic protein p53 rather than Bcl-2 and Bax expression in the melanocytes23, the levels of FLIP and p53 were also examined in the vitiligo epidermis. The result showed that the levels of FLIP were lower and those of p53 were higher in the depigmented epidermis18.
IMPAIRED PI3K/AKT ACTIVATION IN KERATINOCYTE APOPTOSIS
Although more apoptotic keratinocytes occur in the depigmented epidermis, it is unclear how keratinocyte apoptosis occurs in patients with vitligo. In the nonsegmental type vitiligo, depigmented maculopatches could develop on areas corresponding to trauma27, which includes mechanical trauma such as repeated friction28, chemical damage, or ultraviolet (UV) radiation. Although the mechanism of koebnerization is unclear, it may be expected that some inflammation would precede the development of isomorphic depigmentation. In fact, a histological examination of non-segmental vitiligo skin shows prominent inflammatory cells in perilesional areas, which are primarily composed of CD3+, CD4+, and CD8+ T cells29,30, although definite inflammation is rare in patients with vitiligo. A correlation between perilesional T-cell infiltration and melanocyte loss in situ has also been observed31,32. Additionally, there are significantly higher levels of TNF-α, IL-1α, and IL-6 in depigmented than in perilesional, non-lesional, and healthy skin33,34, suggesting that cytokines are important in the depigmentation process as potent inhibitors of melanocyte growth in patients with vitiligo33,35.
TNF-α is a key initiator of inflammation that increased in the depigmented compared to normally pigmented epidermis (Fig. 3). This member of the TNF superfamily of cytokines is at the center of the extrinsic pathway of apoptosis. The extrinsic pathway is triggered by the binding of death ligands of the TNF family to their appropriate death receptors on the cell surface. This, in turn, recruits initiator caspases, giving rise to the pro-apoptotic complex termed DISC. Death receptors (DRs) include TNF-α receptor 1 (TNF-R1), Fas, DR3, DR4 (TNF-related apoptosis inducing ligand-R1; TRAIL-R1), DR5 (TRAIL-R2), and DR6, whereas death ligands include TNF-α, Fas-ligand (FasL), Apo3-ligand, TRAIL, and lymphotoxin36. TNF-α acts through TNF-α receptors. TNF-R1 recruits TNF-R1-associated death domain protein37, which is coupled to two receptors, Fas-associating protein and TNF-R1-associated factor 2 (TRAF2).
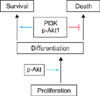
Apoptotic and non-apoptotic signaling occur upon binding of other DRs and ligands. Although more interest have been recently focused on TRAIL and its receptors due to preferential apoptosis of transformed cells and the absence of severe toxicity, the Fas/FasL system is a prototypic inducer of extrinsic cell death (Fig. 4). Therefore, we examined only Fas/FasL in patients with vitiligo. Fas can be activated by FasL, and we found that the expression of FasL and TNF-α increased in the depigmented compared to that in the normally pigmented epidermis38. Fas/FasL leads to recruitment and activation of caspase-8 in the DISC followed by direct cleavage of downstream effector caspases21,39,40. Although intrinsic and extrinsic pathways are separate, there is cross talk between the two pathways. The best characterized connection is the Bcl-2 family member Bid (Fig. 4). In fact, caspase-8 cleaves Bid, generating a truncated form, tBid41,42, which, in turn, translocates to mitochondria and induces the release of caspase-activating factors including cytochrome c from mitochondria, triggering intrinsic apoptosis43. The impact of these two apoptotic pathways could be enhanced when they converge through Bid. In contrast, binding to the TRAF2 activates phosphatidylinositol 3-kinases (PI3K) and its downstream target apoptosis signal-regulating kinase (AKT)44, which, in turn, activates NF-κB45,46 through sequential activation of I-κB kinases and I-κB. These non-apoptotic signaling pathways result in inhibiting apoptosis. Although non-apoptotic signal transduction was induced by the DRs and ligands system such as Fas/FasL, AKT and nuclear factor (NF)-κB activation decreased in the depigmented epidermis (Fig. 5). In addition, inhibiting AKT phosphorylation in cultured normal human keratinocytes treated with TNF-α in the presence of a PI3K inhibitor resulted in a significant decrease in NF-κB phosphorylation with an increase in apoptotic cells. Targeted reduction of AKT1 expression results in significant cell death in the suprabasal layers47, and inhibiting PI3K increases the susceptibility of keratinocytes to apoptosis, with selective death of differentiating keratinocytes (Fig. 6)48. Overall, increased apoptosis of vitiliginous keratinocytes could result from reduced activation of NF-κB via impaired PI3K/AKT activation under increased TNF-α levels46.
REDUCED AQUAPORIN3 (AQP3) DURING APOPTOSIS OF VITILIGINOUS KERATINOCYTES
Activation of PI3K depends on both E-cadherin-dependent cell adhesion and tyrosine phosphorylation events49,50. The intracellular domain of E-cadherin binds directly to β-catenin and γ-catenin. The N-terminal portions of both β- and γ-catenin interact with α-catenin, which links E-cadherin to the underlying actin cytoskeleton. We showed that treatment with the PI3K inhibitor, wortmannin, inhibited E-cadherin-catenin expression and PI3K phosphorylation in cultured keratinocytes51. Cadherin is involved in the calcium-dependent cell-to-cell adhesion of molecules. Increases in intracellular calcium could recruit PI3K to the E-cadherin-catenin complex at the plasma membrane52, resulting in PI3K activation. Because activation of the PI3K/AKT pathway is required for keratinocyte differentiation53, E-cadherin plays a pivotal role in keratinocyte differentiation and cell-to cell adhesion54,55, we previously showed that phosphorylated PI3K decreased in the depigmented epidermis of patients with vitiligo compared to that in normally pigmented epidermis38. Additionally, we identified that AKT1 phosphorylation and E-cadherin, β-catenin, and γ-catenin expression decreased significantly in depigmented epidermis51. We performed a microarray analysis to compare the cultured keratinocytes from normally pigmented epidermis and depigmented epidermis. There was a 2-fold or greater down-regulation of AQP3 expression in the keratinocytes from depigmented epidermis, which was confirmed by Western blot analysis and immunohistochemistry using paired epidermal specimens obtained from 10 patients with vitiligo51. AQP3 has been identified in multiple epithelial tissues as a transmembrane protein localized to the basolateral membrane56. AQP3 is detected in the epidermal keratinocytes below the stratum corneum and is involved in skin hydration57. Although the role of AQP3 in patients with vitiligo is unknown, basolateral membrane AQP3 is delivered to post-Golgi structures directly to form cell-to-cell contacts, where it co-accumulates with E-cadherin58. Activation of PI3K depends on both E-cadherin-dependent cell adhesion and tyrosine phosphorylation events49,50. PI3K/AKT1 phosphorylation and the expression of E-cadherin-catenin complex are reduced in patients with vitiligo51. Therefore, a connection between AQP3 and PI3K through the E-cadherin-catenin complex should be considered. Particularly, the role of AQP3 in keratinocyte apoptosis was examined using cultured normal human keratinocytes with or without transfection of AQP3 small interfering RNA (siRNA)51, because impaired PI3K/AKT activation is involved in vitiliginous keratinocyte apoptosis38. Western blot analysis showed that AQP3 siRNAs in cultured normal human keratinocytes decreased PI3K-p85α phosphorylation significantly, a regulatory subunit of class 1A PI3K, and the expression of E-cadherin, β-catenin and γ-catenin. Immunoprecipitation using PI3K-p85α also showed reduced binding of E-cadherin-catenins to PI3K-p85α51. These results suggest impaired cell-to-cell adhesion. In addition, immunohistochemistry using patient's skin showed that expression of the AQP3 protein, which was localized to the cell borders of viable cells, decreased in depigmented epidermis. Therefore, co-localization of AQP3 and E-cadherin was examined by double staining of cultured keratinocytes and vitiligo epidermal specimens using anti-AQP3 and E-cadherin antibodies. A functional link between AQP3 and E-cadherin was identified as expression of AQP3 and E-cadherin decreased at the cell-to-cell contacts in the depigmented epidermis (Fig. 7A) as well as in the cultured AQP3 knockdown keratinocytes (Fig. 7B)51. A similar E-cadherin staining pattern has been briefly mentioned in lesional, non-lesional, and normal skin59. Inhibiting cadherin increases apoptosis55; therefore, the association between AQP3 knockdown and reduced cell survival was expected. AQP3 knockdown in cultured normal human keratinocytes decreased the number of surviving keratinocytes at low and high calcium concentrations51. Collectively, the reduced AQP3 in vitiliginous keratinocytes could be responsible for their reduced survival51.
ROLE OF KERATINOCYTE APOPTOSIS IN DEPIGMENTATION OF VITILIGO
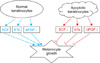
Cell-to-cell interactions play an important role in homeostasis and regeneration of adult tissues. Crosstalk between keratinocytes and melanocytes has been observed. Actually, direct cell-to-cell contact stimulates in vitro proliferation of melanocytes, and growth factors produced by adjacent keratinocytes regulate the proliferation and differentiation of melanocytes60. Keratinocytes produce and release numerous synergistic mitogens in culture, such as basic fibroblast growth factor (bFGF), endothelins, stem cell factor (SCF), hepatocyte growth factor, nerve growth factor, granulocyte macrophage colony stimulating factor, leukemia inhibitory factor, α-melanocyte stimulating hormone, and adrenocorticotrophic hormone61. In particular, SCF is essential for melanocyte survival. Melanocytes express c-kit62, a receptor for SCF63. SCF regulates proliferation and differentiation in neonatal mouse epidermal melanocytes in culture with a cyclic adenosine monophosphate stimulator and bFGF64. SCF exists as membrane-bound and soluble forms65. The proportion of each form is dependent on SCF mRNA, which is either full-length or alternatively spliced and lacking the exon 6 coding region, and on protease activity, which attacks a protease-sensitive site in exon 666. Normal human keratinocytes express both forms of SCF mRNAs67. In our experience, SCF mRNA is maintained as long as sufficient cells are alive, which was more than 3 days in melanocyte culture medium. The number of cultured normal human melanocytes increased in the presence of human keratinocytes and/or SCF, whereas the number decreased in a dose-dependent manner following treatment with an anti-SCF antibody60.
It could be expected that keratinocyte apoptosis reduces these factors for melanocyte growth. Apoptosis of cultured normal human keratinocytes has been induced by UV radiation or treatment of H2O2 or staurosporine. Particularly, staurosporine induces apoptosis in all nucleated mammalian cell types68, regardless of the state of differentiation or the cell-cycle phase. Treatment with staurosporine induced keratinocyte apoptosis of in a dose-dependent manner60. More importantly, SCF mRNA and protein expression decreases with increasing proportions of apoptotic keratinocytes60. We showed previously that more keratinocytes in the depigmented epidermis were apoptotic compared to that in normally pigmented epidermis18,38,51. Therefore, lower levels of keratinocyte-derived melanocyte growth factors, including SCF, were expected in the depigmented epidermis of patients with vitiligo. A Western blot analysis showed that SCF and bFGF protein expression levels were significantly lower in the depigmented epidermis than those in the normally pigmented epidermis of patients with vitiligo60. These results agree with those from an immunohistochemistry-based study that demonstrated significantly lower expression of SCF and endothelin-1 in lesional skin compared with perilesional or nonlesional skin in patients with vitiligo19,33. Withdrawal of the relevant trophic substances results in a rapid decrease in cell number, and apoptosis has been implicated and identified as the mechanism responsible for reducing cell number after withdrawal of the mitogenic stimulus69-71. A reduction in melanocyte growth factors including SCF resulting from apoptotic death of keratinocytes could provide a circumstance for withdrawal of a mitogenic stimulus. Removal of either keratinocyte feeder or exogenous SCF from melanocytes cultured in both results in melanocyte apoptosis60. Overall, keratinocyte apoptosis could induce at least the loss of survival stimuli (Fig. 8), and then passive melanocyte death.
CONCLUSIONS
Vitiligo is not a disease confined to melanocytes. As functional and structural units with melanocytes, keratinocytes in depigmented epidermis may constitute a different microenvironment compared to those in normally pigmented epidermis. These differences could induce less keratinocyte-derived melanocyte growth factor production, resulting in passive melanocyte death with the development of vitiligo.


 XML Download
XML Download