

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Poikiloderma vasculare atrophicans (PVA) is a rare variant of early-stage mycosis fungoides (MF), a malignant neoplasm of T-lymphocyte origin1. Several terms, including "Parakeratosis variegata", "parapsoriasis lichenoides", "lichen variegates", and "parapsoriasis poikilodermique"2, have been used to describe the group of diseases with clinical manifestations similar to those of PVA. Histological findings are poikilodermic and are similar to those seen in long-standing patch or plaque lesions of the classic form of MF3. Immunohistochemical studies also show phenotypic findings similar to those of typical MF1. PVA has been known to show a benign course, without progression to the tumor stage of mycosis fungoides4. However, there is also controversy with regard to whether PVA belongs to the category of an early-stage MF or premycosis2. We report on a case of PVA with dominant CD8+ cytotoxic lymphocytes having the characteristic clinical findings of generalized hyperkeratotic scaly papules with a retiform pattern and a long course of disease without progression to the tumor stage of MF.
CASE REPORT
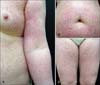
In September 2006, a 59-year-old Korean woman presented with asymptomatic diffuse erythematous scaly macules and hyperkeratotic papules over her entire body, which appeared over a period of 25 years. At first, skin lesions appeared on both thighs. When she visited our department 25 years ago, at the age of 34, we performed a skin biopsy, and recommended treatment with phototherapy. However, she was unable to tolerate the treatment. Subsequently, she had not received any treatment, and the lesions spread slowly over her entire body. Her medical and family histories were non-contributory. Review of systems was negative for rheumatologic and neurologic symptoms. On physical examination, erythematous to violaceous reticulated confluent papules were found to be distributed in a net-like pattern on her anterior and posterior trunk. The upper and lower extremities showed more severe manifestations, especially on the inner sides of their proximal parts (Fig. 1). Laboratory results were within normal limits. Histopathological examination of skin biopsy specimens from the thigh showed compact hyperkeratosis, focal parakeratosis, and epidermotropism of atypical lymphocytes without formation of Pautrier's micro-abscesses in the epidermis, with vacuolar changes in the basal layer. Band-like inflammatory cell infiltrations of mostly lymphocytes with dilated capillaries, and abnormal, wiry patterned collagen bundles were observed in the upper dermis (Fig. 2). In immunohistochemical staining, lymphocytes were positive for CD3, CD45RO, and CD8, and negative for CD4, CD20, CD30, CD56, TIA-1, and granzyme B (Fig. 2). Review of the thigh skin biopsy specimen taken 25 years earlier revealed similar, but less-prominent findings (Fig. 3). A T-cell receptor (TCR) gene rearrangement study of the skin biopsy specimen was performed using a polymerase chain reaction (PCR) technique, and revealed polyclonality. All types of TCR genes, such as TCR-γ, -β, and -δ, were included as PCR targets for clonality testing with DNA extracted from paraffin-embedded tissue. Whole-body computed tomography scans demonstrated several slightly enlarged lymph nodes in both inguinal areas. On the basis of the clinical findings, histopathology, and immunohistochemistry, the patient was diagnosed as PVA. We treated the patient with oral retinoids; however, she refused to continue taking the medication. She has had no further progression over the subsequent 4 years, until now.
DISCUSSION
A wide range of atypical presentations of MF, including erythrodermic, poikilodermatous, verrucous, hyerkeratotic, hypopigmented, vesicular, bullous, and pustular, have been described4. Through the literature review, in 1890, Unna first described the term "parakeratosis variegata (PV)" as lichenoid confluent papules forming a retiform pattern, associated with epidermal atrophy. Similar skin conditions under such different names as parapsoriasis lichenoides, lichen variegates, parapsoriasis poikilodermique, poikilodermal form of mycosis fungoides, or PVA were subsequently reported by several other authors2. According to several recent textbooks, PVA may be seen in three different settings: (a) in association with three genodermatoses; (b) as an early stage of mycosis fungoides; and (c) in association with dermatomyositis, and, less commonly, lupus erythematosus4-6. In a recent review article, Kazakov et al.3 also introduced PVA as poikilodermic MF characterized by alternating hypo- and hyperpigmentation, dryness, atrophy, and telangiectasia. However, controversy remains with regard to whether PVA is a premalignant condition or represents early presentation of cutaneous T-cell lymphoma (CTCL).
Lesions of PVA typically occur on the trunk and flexural areas of middle-aged patients, with a male predilection1. Lesions usually manifest as asymptomatic or mildly pruritic flat-topped, scale-covered papules coalescing into retiform patterns, with net-like or zebra-like distributions, and telangiectasia between papules. A biopsy from poikilodermic areas will show histological findings similar to those seen in long-standing patch or plaque lesions of the classic form of MF3. Our patient's skin lesions initially appeared on both thighs 25 years ago and the lesions spread slowly over her entire body. The asymptomatic, erythematous-to-violaceous, reticulated confluent papules were distributed in a net-like pattern on her anterior and posterior trunk. Observance of characteristic clinical manifestations and histological findings resulted in a diagnosis of PVA. We herein report on a 59-year-old Korean woman with PVA who showed some remarkable features, as follows.
First, she has had a long-term benign course of PVA for approximately 30 years without progression to the tumor stage, even though she has not received any treatment. Histopathological findings showed some progression, compared with the specimen taken 25 years ago, but were still in the late patch stage or the early plaque stage. In recent literature, Kreuter et al.7 reported on a case of PVA that remained in the patch stage for 25 years, and showed a band-like superficial lymphocytic infiltrate consisting mainly of CD4+ cells and a negative TCR gene rearrangement. To the best of our knowledge, three cases involving similar diseases have been reported in the Korean literature as "Parapsoriasis Variegata" in 19798, as "Poikilodermatous MF" in 19999, and as "PVA" in 199910. However, all patients were lost to follow-up, and TCR gene rearrangement tests were not performed.
Second, immunohistochemically, lymphocytes were positive for CD3, CD45RO, and CD8, and negative for CD4 in our patient. The typical phenotype of the classic form of MF presents CD3+, CD4+, CD45RO+, and CD8-, with common loss of CD7. However, there have been some case reports of the CD8+ phenotype, namely, a CD8+ cytotoxic variant of MF. They are rare and their clinical behavior is not well defined11. A more aggressive clinical course has been noted in some cases12,13, whereas a clinical and histological similarity to the classic CD4+ MF has also been reported14. We agree with the opinion of Nikolaou et al.15, who presented a series of seven cases of CD8+ MF. They reported that special clinical features, such as hyperpigmentation and poikiloderma, were often noted in CD8+ phenotypes of MF and their long-standing, indolent courses suggested that CD8 may represent a marker of mild biological behavior. Our patient showed a CD8+ dominant phenotype, which could explain the long, benign clinical course over a period of 30 years.
Third, TCR gene rearrangement study using all types of PCR genes revealed polyclonality in our patient. According to the literature, TCR-beta- or gamma-chain genes show monoclonal rearrangement in most cases of MF, including patch-stage disease3. Although PCR-based TCR gene rearrangement has been reported to show great sensitivity and specificity for T-cell lymphoma in recent studies, there were also several cases in which clonality could not be detected16,17. The reasons for the negative result in our case may be due to limitations of methodology, such as preparative process of small samples (4-mm punch biopsy specimens), PCR techniques, or early-stage of the disease. In addition, detection of a monoclonal T-cell infiltrate is not lymphoma specific18; therefore, monoclonality does not always equal malignancy16. Thus, despite the clear association with CTCL, we think that monoclonality is not a prerequisite for diagnosis of PVA, and that clinical presentation and histopathology are major factors determining the diagnosis of early-stage MF. On the contrary, Kikuchi et al.2 contended that conditions having a high risk of malignant transformation to lymphoma but without detectable evidence of monoclonality should be considered as "premycosis", although a positive TCR gene rearrangement is not absolutely required for the diagnosis of malignant lymphoma.
In conclusion, the question remains as to whether our case of PVA corresponds to a premycotic condition or early-stage MF; however, it is meaningful that PCR detection of TCR gene rearrangement may be useful in monitoring progression of PVA to advanced stage of MF. The CD8+ immunophenotype of MF and polyclonal rearrangement of TCR genes were thought to reflect the long-benign course of our patient. Regular long-term follow-up with repeated TCR gene rearrangement study will be needed for monitoring of disease progression.


 XML Download
XML Download