

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Dermatomyositis (DM) is a rare multisystemic non-suppurative inflammatory disease of the skeletal muscles, characterized by typical skin lesions, and symmetric proximal extensor myopathy. Calcinosis cutis is frequently accompanied by juvenile dermatomyositis (JDM). Ulceration may be observed in the lesions of calcinosis cutis1,2. Cases of the co-existence of ulcerated calcinosis cutis and adult DM have been rarely reported3,4. The current case is presented due to the relatively rare incidence of ulcerated calcinosis cutis in patients with adult DM, as well as the co-existence of vulvar lichen sclerosus (LS), which has not been reported previously to the best of our knowledge.
CASE REPORT
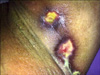
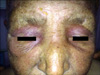
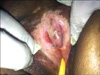
A 71-year-old woman was admitted to our hospital with complaints of wounds in both axillae and below the right breast over the past two months, as well as violet-colored edematous erythema on the upper eye lids, difficulties in walking, sitting, and lifting of the arms for the past 6 months, dysphagia, and inability to urinate for the past 45 days. The patient underwent surgery for stage IV adenocarcinoma of the ovary, and had a history of type-II diabetes mellitus. Dermatologic examination revealed ulcerated-nodular lesions in the bilateral axillary and right inframammary regions (Fig. 1), hard nodular lesions in the bilateral pectoral muscles, bilateral violet-colored erythematous and edematous eyelids (Fig. 2), violet-colored/hyperpigmented patches on the knees and elbows, and poikilodermatous eruptions on the anterior surface of the chest, scapular area and shoulders. An eroded, atrophic, depigmented patch was also observed on the labium minus and clitoris (Fig. 3).
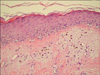
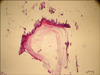
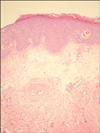
Histopathologic examination of a punch biopsy obtained from the patient's poikilodermatous lesions demonstrated scattered vacuolar degeneration of the thin layer of orthokeratosis at the base of the epidermis, mild edema in the upper dermis, melanophages, and perivascular lymphocytic infiltration (Fig. 4). Histopathologic analysis of the excisional biopsy obtained from the nodular lesions in the pectoral region demonstrated a perivascular chronic inflammatory cell infiltration with papillomatous appearance in the dermis and extensive lamellar calcification in the subcutaneous adipose tissue (Fig. 5). Histopathologic analysis of a punch biopsy taken from the depigmented lesions of the vulva revealed a thin layer of lamellar hyperkeratosis with scattered atrophy, scattered acanthosis, papillomatous, scattered edema in the upper dermis, and patchy collagen deposition in the dermis. In addition to dilated and congested vessels, a mixed inflammatory cell infiltration was also observed (Fig. 6). These findings were consistent with DM, calcinosis cutis and LS, respectively.
The esophageal passage graph performed to evaluate the dysphagia was normal. No pathologic findings were observed in the cranial, cervical, abdomino-pelvic magnetic resonance imaging, thoracic computed tomography, mammography, and breast ultrasonography (USG), which were performed due to a history of malignancy.
The electromyography performed to investigate muscle weakness demonstrated findings of neuropathy and myopathy. The results of the direct urinary system graphy and abdominopelvic USG were within normal limits. The hemogram was consistent with the anemia of chronic disease. The aspartate aminotransferase, alanine aminotransferase, creatine phosphokinase, calcium (Ca), and phosphate (P) levels were normal, whereas the lactate dehydrogenase level was high. The result of the antinuclear antibody test was negative. The patient was started on systemic prednisolone (60 mg/day), colchicine (1.5 g/day), and hydroxychloroquine (400 mg/day). Topical treatment for the ulcerated area included a collagen-based wound dressing, pomade including collagenase, fusidic acid cream, and clobetasol 17-propionate cream for the vulva. On the 3rd week of treatment, some of the ulcerated lesions regressed with epithelization, and lesions on the trunk and extremities almost completely healed with hyperpigmentation. Moreover, the edema and violet-colored erythema on the face faded mildly, and the nodular lesions significantly regressed. The treatment was continued for 6 weeks, but no improvement was observed in the lesions of the vulva. By the end of the treatment period, partial regression was observed in the patient's skin lesions, but no improvement was reported, with difficulties in swallowing and muscle weakness. Total parenteral nutrition solution and enteral nutrition by nasogastric tube were initiated by the end of the 4th week due to progressive difficulty in swallowing. The patient was being monitored for recurrent catheter-related infections, blood glucose level and electrolyte imbalance, and died during the 6th week of treatment.
DISCUSSION
DM is an autoimmune connective tissue disorder characterized by typical skin lesions and symmetric idiopathic inflammatory myopathy. The disease affects both children and adults, and is more frequently encountered in women than in men. Violet-colored papules and plague spread over bony protrusions on the dorsa of the hands (Gottron's papules), and symmetric, violaceous erythematous macules on the knees and elbows (Gottron's sign) are pathognomonic of DM. Other skin lesions include periorbital violaceous (heliotrope) erythema/edema, periungual telangiectasia, erythema in a photosensitive distribution, poikiloderma, and alopecia. In our patient, we observed periorbital violet-colored erythema and edema, violet-colored/hyperpigmented patches on the knees and elbows and poikilodermatous eruptions on the anterior surface of the chest, scapular areas, and shoulders1-4.
In addition to skin involvement, DM is a systemic disease with skeletal muscle involvement, characterized by symmetric, proximal, and extensor myopathy, and it can be accompanied by arthritis, arthralgias, esophageal disease, or cardiopulmonary dysfunction4. Our patient also presented with dysphagia associated with esophageal involvement, and difficulties in walking, sitting, and lifting of the arms associated with proximal extensor myopathy.
The relationship of DM with cancer is well-known. The incidence of internal malignancy in patients with adult DM has been reported to range from 10~50%. The most frequently reported malignancy is ovarian cancer, followed by genitourinary malignancies and colon cancer4,5. Our patient had a history of stage IV adenocarcinoma of the ovary, which had been diagnosed 4 years ago. The patient underwent surgery for malignancy and received six courses of chemotherapy.
The treatment of cutaneous lesions of DM involves the use of sun protection agents, topical corticosteroids, systemic hydroxychloroquine, quinacrine, low-dose methotrexate, and retinoids, whereas immunosuppressive agents, such as systemic steroids, methotrexate, azathioprine, intravenous immunoglobulin, cyclophosphamide, and cyclosporine are used for the treatment of muscle weakness and systemic findings1. Our patient was also started on combined corticosteroid therapy and hydroxychloroquine treatment.
LS is a rarely encountered chronic inflammatory disease of the skin and mucosal surfaces, which most commonly involves the anogenital region. The exact cause of LS is unknown. Familial occurrence of LS suggests that LS may have a genetic link. LS is clinically characterized by polygonal papules, and porcelain-white plaques, with atrophic fragile skin, fissures, erosions, telangiectasias, erythema, and different degrees of sclerosis in the anogenital region. It can also be observed in the other regions of the body. LS is most frequently observed in elderly women, although it can be seen in any age group6,7.
Dyspareunia, urinary tract obstruction, constipation, infections secondary to ulceration and the development of squamous cell carcinoma associated with LS are the primary complications observed in women. The first choice of treatment is the application of ultrapotent topical corticosteroids6,7. In our case, the complaint of inability to urinate was considered to be due to vulvar LS. Topical clobetasol 17-propionate cream was used for the vulva lesions.
Calcinosis cutis is an important complication of DM, characterized by the accumulation of hydroxyapatite crystals and amorphous calcium phosphate in the skin and soft tissue8. Although calcinosis cutis is mostly encountered in JDM, it is rare in adult onset chronic DM9,10. Muller et al. reported the incidence of cutaneous calcinosis as 20% and 74% among adults and children with DM, respectively11. Cases of the co-existence of ulcerated calcinosis cutis and adult DM have been rarely reported12,13. Calcinosis cutis appears as a late symptom and dystrophic calcifications are observed11,14. Calcinosis cutis may sometimes lead to severe pain with marked weakness, joint contractures, persistent infectious ulceration of the skin, cosmetic disorders9, and muscular atrophy. In adults, lesions are frequently in the form of dermal or subcutaneous nodules and are usually observed in the sites of repeated microtrauma, such as the elbows, knees, and other acral areas12,15. Large subcutaneous tumoral deposits may also form on the trunk of the body4. The risk factors involved in the development of calcinosis cutis include delayed treatment and severity of the course of disease16. In our case, ulcerated nodular lesions were seen in both bilateral axillary and right inframammary regions, as well as in bilateral pectoral regions. Our patient showed relatively early manifestation of calcinosis cutis, which may be related to delayed treatment and severe course of the disease. The patient was started on colchicine (1.5 g/day).
Spontaneous regression has been reported in some patients with calcinosis; however, no effective treatment modality has yet been suggested. On the other hand, various degrees of success have been reported with bisphosphonates, probenecid, warfarin, aluminum hydroxide, colchicine, diltiazem, surgical excision, and intralesional steroid either as monotherapy or in combination, depending on the clinical circumstances9,13,15,17-19.
Prednisolone (60 mg/day), colchicine (1.5 g/day), and hydroxychloroquine (400 mg/day) were administered to the patient with severe systemic and cutaneous symptoms. Moreover, topical treatment with a collagen-based wound dressing that was a pomade including collagenase and fusidic acid cream, was applied to the ulcerated areas. We observed that some of the ulcerated nodular lesions regressed with epithelization, while skin lesions on the face, trunk, and extremities significantly regressed. There are studies reporting successive results in the treatment of cutaneous and muscle weakness symptoms of DM with steroid therapy within one to several months, suggesting the maintenance of steroid therapy for 6 months to 2 years through dose reduction1,2. Various case reports have demonstrated that regression of calcinosis cutis co-existing with juvenile or adult DM and localised linear scleroderma is observed within a mean period of 4 months with colchicine treatment, over 2.5 years with diltiazem treatment, 8 months with aluminum hydroxide treatment, and 2 years with hydroxychloroquine, azathioprine and prednisolone treatments12,18-20. In our case, the patient died due to secondary infections and metabolic disorders, we could not perform long-term monitoring to observe a complete clinical response to systemic therapy.
Previous reports have documented that DM/polymyositis may be associated with other autoimmune connective tissue diseases such as systemic lupus erythematosus, scleroderma, rheumatoid arthritis, Sjögren's syndrome and inflammatory, dermal and subcutaneous connective tissue disease such as morphea profunda1,21.
Here, we demonstrated the co-existence of ulcerated calcinosis cutis in a patient with adult DM, as well as the co-existence of vulvar LS, which was the first reported case to the best of our knowledge. The coexistence of both diseases may be incidentally, inevitably or immune mediated. In our case, there are two possible explanations of the relationship between DM and LS. First, DM patients have some autoantibodies resulting from an immunemediated process. DM patients have antisynthetase antibodies which are often associated with overlap syndromes. IgG autoantibodies against extracellular matrix protein 1 are involved in the pathogenesis of LS. A likely relationship with autoimmunity is also possible. LS may be associated with the HLA-DQ7 region of MHC class II antigen, which is related to an increased risk of autoimmune diseases. Secondly, a 19% overlap rate has been reported in adult DM cases with other autoimmune connective tissue diseases. The most frequent overlap in adult DM is scleroderma. A coexistence with DM and morphea profunda has also been reported21. LS is known to be closely related to morphea. Scleroderma, morphea and LS are a group of diseases which are associated with dermal sclerosis1,21,22.
Even though the exact relationship between DM and LS is not apparent, this case report presents a rare connective tissue disease which affects the upper dermis and epidermis, which may coexist with DM.
 XML Download
XML Download