

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Castleman's disease (CD) is an angiofollicular lymphoid hyperplasia, or giant lymph node hyperplasia. It is characterized by lymph node hyperplasia and vascular proliferation1,2. It was first described in 1956 by Castleman et al.3 as a localized lymphadenopathy that usually involves mediastinum or pulmonary hilus resembling thymoma. Keller et al.2 divided CD histologically into three types: hyaline-vascular, plasma cell, and mixed type. Several associated diseases of CD have been described, including paraneoplastic pemphigus4, thrombotic thrombocytopenic purpura5, amyloidosis6, and Kaposi sarcoma7. A few cases of CD with cutaneous involvement have been reported8-15. However, there are no reports in the Korean literature. Here, we report a case of CD which showed a characteristic cutaneous manifestation of multiple violaceous plaques with multiple lymphadenopathy that responded well to chemotherapy.
CASE REPORT
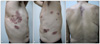
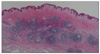
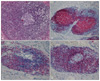
In May 2008, a 65-year-old man presented with multiple violaceous plaques which had developed over the previous 7 years. Lesions started as small violaceous macules on left axilla, and then spread to the scalp, trunk, and legs. They slowly formed confluent indurated plaques. He had pulmonary tuberculosis and tuberculosis lymphadenitis which were diagnosed by open lung biopsy 4 years prior and had been treated with oral medication. His family history was non-contributory. On physical examination, variable sized reddish to violaceous indurated plaques were observed on his scalp, chest, back, axilla, and both legs (Fig. 1). Superficial lymph nodes were palpable in his neck, axilla, and inguinal areas. A previous operative scar was observed on his right side of chest. He has been suffered from night sweats for several years. However, there were no other systemic symptoms such as fever, weight loss, or asthenia. On laboratory examination, we found anemia (Hgb 9.4 g/dl, normal range: 13.5~17.5), thrombocytosis (platelet 555×103/mm3, normal range: 150~350×103), increased erythrocyte sedimentation rate (ESR) (105 mm/hr, normal range: 0~17), hyperproteinemia (total protein 11.4 g/dl, normal range: 5.5~8.0), and polyclonal hypergammaglobulinemia (IgG 4,030 mg/dl, normal range: 614~1,295; IgM 547 mg/dl, normal range: 53~334). Hepatitis B antigen and anti-human immunodeficiency virus (HIV) antibody were negative. A peripheral blood smear showed a mild degree of microcytic hypochromic anemia with rouleaux formation and thrombocytosis without definite abnormal cells. A bone marrow biopsy revealed a small increase of mature plasma cells (approximately 7.3%). There were multiple hypermetabolic lymph nodes on his neck, chest, and abdomen. Splenomegaly was determined by positron emission tomography-computed tomography but there was no other internal solid organ involvement. An excisional biopsy from the left inguinal lymph node showed follicular formation with many lymphocytes and dense plasma cells (Fig. 2). A skin biopsy taken from a right flank lesion revealed variable sized lymphoid follicles in the dermis (Fig. 3) and nodular infiltrates of lymphocytes, lymphoplasmacytoid cells, and many plasma cells (Fig. 4A). In the immunohistochemical stain, lymphocytes and plasma cells stained positively for CD 20, CD 79a, CD 5, CD 45RO, and bcl-2 (Fig. 4B~D). CD3 was weakly positive in the reactive T cells. CD10 and bcl-6 were negative. Kappa and lambda light chain were equally stained.
These findings were consistent with multicentric plasma cell type of CD. He was treated with 8 cycles of CHOP chemotherapy (cyclophosphamide, adriamycin, vincristine, and prednisone). The size and hypermetabolic activities of lymph nodes decreased and skin lesions were much improved with mild hyperpigmentation. He remains healthy 2 years after treatment.
DISCUSSION
CD is an uncommon B-cell lymphoproliferative disorder. The time to diagnosis the disease is often long, which is attributable to clinical polymorphism and poor awareness of the disease. CD can occur at any age, even during childhood, with peak frequency during adulthood1. There are two clinical forms of CD: unicentric and multicentric2. The onset age of multicentric CD is older than that of unicentric CD. There are no gender differences in unicentric CD but males have multicentric CD more often than females1,9. The unicentric (solitary) form commonly presents as a mass in mediastinum. It can also be found outside the thorax (both nodal and extranodal), for example on the neck, arm, abdomen, vulva, and pericardium1,16. The multicentric form shows generalized lymphadenopathy and various systemic symptoms. It may also be associated with POEMS syndrome1,17.
Histologic variants of CD are composed of hyalinevascular (90%), plasma cell (10%), and a mixed type2,12. The hyaline-vascular type represents 80 to 90% of all cases of the solitary form. The multicentric form consists almost entirely of plasma cell type18. The hyaline-vascular type lacks the systemic signs and/or symptoms which are commonly associated with plasma cell types17. Histopathologically, it is characterized by small, concentrically whorled, lymphoid follicles surrounded by small lymphocytes arranged in a concentric, onion-skin pattern. Extensive capillary proliferation and hyalinized sclerotic collagen are present between follicles9. The plasma cell type shows many systemic signs and/or symptoms including fever, anemia, weight loss, loss of appetite, elevated ESR, hypergammaglobulinemia, and hypoalbuminemia18. It reveals as well developed hyperplastic secondary lymphoid follicles and many plasma cells exist predominantly in the interfollicular areas13. Occassionally, there are numerous Russell bodies, deposition of an amorphous acidophilic material that probably contains fibrin, and immune complexes in the center of follicles1.
Cutaneous involvement of CD has rarely been reported in the multicentric form. Among ten reported cases8-15, three patients were male and seven patients were female. Their mean age was 50.8±12.7 yrs (range: 36~72 yrs). All skin lesions manifested as multiple erythematous to brownish nodules and plaques predominantly on the back but also on the face, trunk, and extremities. Histologic features were plasma cell type except in one case. Results are summarized in the Table 1. In our case, the patient showed multiple reddish to violaceous indurated patches on his scalp, chest, back, axilla, and both legs, which is similar to other cases. A skin biopsy and a lymph node biopsy showed nodular aggregates of many lymphocytes and plasma cells with Russell bodies in the dermis. Immunohistochemical staining for kappa and lambda light chains confirmed the polyclonal nature of the plasma cell infiltrate. These findings were compatible with plasma cell type CD.
The pathogenesis of CD is still not understood. Interleukin (IL)-6 is an important factor in the pathogenesis of CD, especially in the multicentric form12. IL-6 contributes to proliferation and maturation of B lymphocytes into mature plasma cells and proliferation of blood vessels19. Yoshizaki et al.20 showed that the cells present in the germinal center of the enlarged lymph nodes produce IL-6 and there is close relationship between the serum levels of IL-6 and the clinical symptoms of patients with CD. Recently, the incidence of CD in HIV patients has increased10. Epidemiological and polymerase chain reaction studies have suggested that human herpes virus (HHV)-8 is associated with multicentric CD in all HIV positive patients and in almost 50% of HIV negative patients21,22. HHV-8, known as Kaposi sarcoma associated herpes virus, also produces a functional analogue of IL-6. Although the exact pathogenesis of CD is unknown, immune dysregulation of IL-6 may play a key role10. In addition, CD is associated with high levels of IL-10, tumor necrosis factor-β, interferon-γ, vascular endothelial growth factor, and macrophage colony-stimulating factor11. In our patient, we could not check IL-6 expression or HHV-8 DNA sequences.
The differential diagnosis of CD includes angiolymphoid hyperplasia with eosinophilia (ALHE), Kimura disease, sinus histiocytosis with massive lymphadenopathy, cutaneous plasmacytoma, angioimmunoblastic T cell lymphoma, primary cutaneous marginal zone lymphoma, and primary cutaneous follicular center cell lymphoma10,15. CD can be distinguished from these diseases by combined generalized lymphadenopathy, clinical manifestations, type and patterns of infiltrate cells, and immunohistochemical stain, especially polyclonal restriction of kappa and lambda light chains.
The treatment differs depending on clinical types. Surgical excision is effective for unicentric CD17. This treatment allows full recovery without relapse in almost all cases. On the other hand, there is no consensus for the treatment of the multicentric type. A variety of approaches, including surgery, chemotherapy, radiation therapy, and anti-viral therapies, have been attempted17,18. The treatment of choice is a monthly combination chemotherapy such as CHOP (cyclophosphamide, vincristine, doxorubicin, prednisone) or AVBD (etoposide, ifosfamide, cisplatin alternating with doxorubicin, vincristine, bleomycin and dacarbazine)18. Other therapies, such as interferon-α, retinoid acid or rituximab or anti-IL-6 antibodies, have been used and gave promising results12,23,24. Generally, the prognosis of multicentric CD is poor, but our patient had 8 cycles of CHOP chemotherapy and responded well to treatment.
In conclusion, we report a rare case of cutaneous involvement of multicentric CD which had a good prognosis. However, further long-term follow up for detection of recurrence is needed.


 XML Download
XML Download