

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Benign cephalic histiocytosis (BCH) is a rare, non-Langerhans histiocytosis first described by Gianotti et al1. in 1971. It is characterized by asymptomatic eruptions of slightly raised, round or oval, orange-red, or red-brown papules distributed mainly on the head, face, neck, and shoulders of infants and young children, which shows spontaneous regression with time. The histopathological features show a well-circumscribed histiocytic infiltrate within the superficial to mid-reticular dermis. The histiocytes express CD68 and HAM56, but are characteristically S-100 and CD1a negative.
CASE REPORT
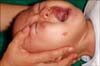
A 7-month-old boy visited our clinic with the complaint of small papules and plaques on his right cheek since the age of 4 months. He had no significant medical history and had normally reached all of his developmental milestones. On physical examination, erythematous grouped papules on wheal-like plaques were observed on the right cheek, as shown in Fig. 1. The laboratory work-up revealed the following: WBC, 125,000/mm3 neutrophil, 25.3% (40~74%); eosinophil, 9.5% (0~7%); and aspartate aminotransferase(AST), 52 IU/L. Results of other laboratory workups were normal.
A biopsy specimen of the upper lesion of a wheal-like plaque showed a spread that infiltrated the entire dermis, with no epidermotropism (Fig. 2A). Most of the cells within the infiltrates were large, epithelioid histiocytic cells with eosinophilic cytoplasm (Fig. 2B). Lymphocytes and eosinophils were scattered among the histiocytes. Immunohistochemistry showed positive staining for CD68 in lesional cells (Fig. 3A), but negative for S-100 protein or CD1a (Fig. 3B, C). Based on these clinical and histological features, we diagnosed this disorder as a benign cephalic histiocytosis. In view of the self-healing nature of the illness, we only observed the patient, without administering any medication. The papules and plaques have gradually become flat and have shown regression for 11 months, leaving brown-pigmented lesions.
DISCUSSION
BCH is a rare non-Langerhans cell histiocytic childhood disorder that presents as a self-healing eruption. The eruption usually starts during the first 3 years of life. Its clinical manifestations are small, red-to-yellow papules distributed mainly on the head, face, neck, and shoulders, although it may become generalized. Cases involving fused papules and formation of reticulated patterns have also been reported3,4. The case discussed in this report displayed clinical characteristics of BCH with papules and plaques on the right cheek. Typically, there is no mucous membrane, acral, or visceral involvement. Spontaneous regression of the eruption is the rule; complete regression occurs within 50 months, on average. However, recent studies have suggested an association of BCH with other non-Langerhans cell histiocytosis disorders, and BCH is now regarded as a part of non-Langerhans cell histiocytosis in its clinical spectrum5-8. In general, BCH does not lead to abnormalities in laboratory test results, or have known complications. However, diabetes inspidus and insulin-dependent diabetes mellitus have been reported in association with BCH from infiltration of the pituitary stalk, which has been described more commonly in Langerhans cell histiocytosis and xanthoma disseminatum9,10.
The histopathologic hallmark of BCH is a well-circumscribed histiocytic infiltration of the superficial to midreticular dermis with attenuation of the adjacent epidermis and flattened rete ridges. Epidermotropism is not a feature, and there is a mixed inflammatory infiltrate composed of lymphocytes and rare eosinophils. Histiocytes are typically large, with abundant eosinophilic cytoplasm and oval, hyperchromic, or vesicular nuclei, often with very prominent nucleoli. Older lesions may contain giant cells, and mitoses are absent. Immunohistochemically, Langerhans cell markers, CD1a and S-100 protein antibodies, are negative, whereas macrophage/ histiocytic markers, CD68 and factor XIIIa, are positive. Histiocytes that had infiltrated the dermis were observed in the histological findings of the case. Results of immunohistochemical staining were negative for CD1a and S-100 and positive for CD68, indicating that BCH belongs within a spectrum of non-Langerhans cell histiocytosis.
Clinical and/or histological differential diagnosis includes the micronodular form of juvenile xanthogranuloma (JXG), Langerhans cell histiocytosis (LCH), and generalized eruptive histiocytosis (GEH)3,5,11. The micronodular form of JXG presents more disseminated eruptions than BCH usually does, and there can be ocular involvement. Histology shows lipid accumulation within histiocytic cells, abundant foamy cells, and Touton giant cells. GEH is microscopically very similar to BCH, but typically affects adults with more extensive distribution of lesions, and occasional mucosal lesions. LCH preferentially affects flexor surfaces with a more florid, scaling, and crusting papular eruption. One or more visceral organs may be involved, and there may be associated fever, malaise, and failure to thrive. Immunohistochemically, LCH differs from BCH in positive staining for CD1a and S-100, and negative staining for CD68 and factor XIIIa. The cases were diagnosed as BCH, based on skin lesions and their distribution characteristics, absence of an association with systemic diseases, conditions that regressed with elapsed time, histological findings that displayed proliferation of histiocytes in the dermis, and immunohistochemical staining findings that indicated traits of non-Langerhans cell histiocytosis [CD1a/S-100 (-O/-), CD68(+)].
Etiology of BCH is unclear; however, there is evidence to support the hypothesis that BCH, GEH, and JXG are part of an overlapping clinical spectrum of disease. Transformation of BCH lesions into JXG has been reported in 2 infants, aged 6 months6 and 1 year7, respectively. In addition, Sidwell et al. reported a case of simultaneous occurrence of disseminated JXG and BCH8. These findings can be interpreted as being supportive of the hypothesis that BCH and JXG are different morphologic expressions of the same disease. BCH, GEH, and the mononuclear JXG share a common histiocytic cell phenotype, notably the vacuolated histiocyte. The fact that the three disorders cannot be differentiated solely based on a histological study also supports such a hypothesis12.
BCH is a self-limiting disorder that shows improvement within several months or years. Thus, distinguishing it from other diseases is important for an accurate diagnosis, and the nature of BCH should be explained to the patient and the family. Although no cases of BCH in association with systemic diseases have been reported, rare cases of diabetes inspidus or insulin-dependent diabetes mellitus in BCH patients, as well as transitions to other non-Langerhans cell histiocytosis disorders, have been reported. Therefore, periodic monitoring of patients'progress would be advisable.


 XML Download
XML Download