

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Eosinophic fasciitis (EF) is an uncommon connective tissue disease, first described in 1974 by Shulman1. It is characterized by scleroderma-like cutaneous changes, peripheral eosinophilia, hypergammaglobulinemia and elevated erythrocyte sedimentation rate (ESR). The absence of Raynaud's phenomenon or sclerodactyly distinguishes EF from scleroderma. Typical histologic findings include chronic inflammatory infiltration affecting deep fascia with lymphocytes, histiocytes, and occasionally eosinophils1. The etiology and pathogenesis of EF remain to be elucidated. This report describes two cases of EF developed after excessive physical activity. They were confirmed by both deep wedge biopsy of the skeletal muscle, including the fascia, and magnetic resonance imaging (MRI), and were managed successfully with high dose corticosteroids.
CASE REPORT
Case 1
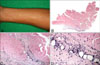
A 36-year-old man presented with a 2-month history of brownish tender induration on both forearms (Fig. 1A). He had experienced immobility of both wrists. There was no history of sclerodactyly, Raynaud's phenomenon, dysphagia, dyspnea nor L-tryptophan ingestion. Chest X-ray was unremarkable. Laboratory investigations revealed white blood cell count at 6,800/mm3, with 8.1% eosinophils. ESR, liver, renal and thyroid function tests were normal. Uric acid level was 7.7 mg/dl (normal range, 2.4~7.0 mg/dl). Serology was traced for anti-nuclear antibody with mixed pattern of speckled and anticytoplasmic antibody, and was negative for anti-ds DNA antibody, anti-Scl-70 antibody, anti-centromere antibody and rheumatoid factor.
Deep tissue biopsy demonstrated fibrotic fascia with mixed infiltrate composed of lymphocytes, histiocytes, and plasma cells (Fig. 1B~D).
EF was diagnosed based on typical skin sclerosis, histopathology results and eosinophilia. He was treated with 60 mg prednisolone daily. Within 1 week, the induration softened and he experienced mildly improved joint mobility. Prednisolone dosage was reduced progressively until complete remission was achieved 2 months following diagnoses. The patient currently remains asymptomatic.
Case 2
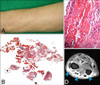
A 52-year-old female presented with symmetrical painful swelling and skin induration on both forearms for 4 months (Fig. 2A). Physical examination revealed marked tenderness of both forearms without evidence of muscle atrophy and weakness. There was no history of sclerodactyly, Raynaud's phenomenon, dysphagia, dyspnea nor L-tryptophan ingestion. Chest X-ray was normal. Hematologic evaluation demonstrated white blood cell count at 6,700/mm3 (eosinophil 7.7%), ESR at 22 mm/hr (normal range, 0~15 mm/hr), IgG 1,841 mg/dl (normal range, 700~1,600 mg/dl). Anti-nuclear antibody test was positive (titer 1:100, weak unspecified), anti-histone antibody was positive (93 U/ml, [normal range, 0~39.9 U/ml]) and anti-ds DNA antibody was negative.
Histologic examination of a deep wedge biopsy taken from the left forearm revealed thickened fascia with infiltration of lymphohistiocytes, plasma cells and hyaline degeneration (Fig. 2B, C). MRI scan revealed increased signal intensity in the fascia and tendon sheath (Fig. 2D).
Treatment with oral prednisolone was started at 60 mg daily initially. Two months later, the eosinophil count was returned to normal. The edematous changes and skin induration of both forearms gradually improved. The dosage of prednisolone was tapered over 5 months and the patient remained asymptomatic.
DISCUSSION
EF is characterized by initially edematous and erythematous extremities, followed by 'peau d'orange' and woody skin induration1. Patients have a variety of non-specific features including malaise, weakness, fever and weight loss. Laboratory investigations may reveal peripheral eosinophilia, hypergammaglobulinemia (usually IgG) and elevated ESR. Anti-nuclear antibodies, rheumatoid factor and anti-ds DNA antibodies may be present. It occurs equally in males and females, and most patients are in their third to sixth decades2. Pediatric disease, however, has also been documented3. EF has been frequently associated with hematological disorders such as aplastic anemia, hemolytic anemia, peripheral autoimmune thrombocytopenia, pernicious anemia, leukemias, and lymphomas4,5. Some autoimmune diseases such as Hashimoto's thyroiditis, morphea, systemic sclerosis, Sjogren's syndrome and antiphospholipid antibody syndrome have also been reported in patients with EF6.
The etiology and pathogenesis are unknown. At least 66% of patients relate the onset of the disease to previous excessive physical activity7,8. In the two cases discussed above, there had been a history of vigorous physical activity (weight training [case 1], and excessive housework [case 2]). Blaser et al.8 suggested that excessive exercise or trauma may trigger antigenicity of the fascia and subcutis. Other hypothesized triggers include trauma9, phenytoin10, trichloroethylene11 and Borrelia burgdorferi infection12. EF is confirmed by histopathologic findings. Typical findings are: thickening of the fascia associated with inflammatory cell infiltration consisting of lymphocytes, plasma cells, histiocytes, and eosinophils in variable degrees. The presence of eosinophils in the fascia is not confirmatory of the diagnosis7,13. Additionally, Baumann et al.14 suggested that MRI in EF demonstrates typical findings, including abnormal signal intensity, thickening and contrast enhancement of the fascia. Therefore, MRI can facilitate diagnostic investigations, guide biopsy site and evaluate therapeutic response.
The differential diagnosis of EF includes morphea, systemic sclerosis and eosinophilia-myalgia syndrome. In morphea, the inflammatory process predominantly affects the reticular dermis and superficial subcutis, but EF is characterized by involvement of the deep subcutis and fascia7. The epidermis, papillary dermis and adnexa are not affected13. In contrast to systemic sclerosis, sclerodactyly and Raynaud's phenomenon, internal organ involvement are absent in EF. Eosinophilia-myalgia syndrome has similar findings, both clinically and histologically, with EF, but has a history of L-tryptophan ingestion, polyneuropathy and pulmonary symptoms13. Our patients have classical EF histories and investigation outcomes: the onset was rapid after vigorous physical activity, with peripheral eosinophilia and hypergammaglobulinemia. They did not experience sclerodactyly, Raynaud's phenomenon, dysphagia, dyspnea and L-tryptophan ingestion. Their epidermis, papillary dermis and adnexa were not affected. The fascia of both patients showed thickening or fibrosis with inflammatory cell infiltration.
The gold standard of EF treatment is high-dose corticosteroids, reported to be effective in up to 70% of cases7. Other therapeutic regimens include hydroxychloroquine, ketotifen, cimetidine, methotrexate, cyclosporine, penicillamine, azathioprine and griseofulvin15, but patient response has been limited. Recently, Weber et al.16 reported that a combination regimen, consisting of UVA1 phototherapy, isotretinoin and corticosteroid, led to good clinical outcomes. The supposed effects of isotretinoin are inhibition of fibroblast growth and reduction of collagen production in dermal fibroblasts. The likely effects of UVA1 phototherapy on matrix metalloproteinase-1, and that of prednisone as an anti-inflammatory effect make this combination regimen an attractive option for managing EF, requiring lower steroid dosage and treatment time.
The clinical outcome of EF is usually good, except when it is associated with a malignant hematologic disease. Endo et al.17 suggested that young age (under 12 years) at onset, trunk involvement and morphea-like skin sclerosis might be a hallmark of refractory disease. Jinnin et al.18 reported that EF patients have significantly higher serum tissue inhibitor of metalloproteinase-1 (TIMP-1) level than does a healthy person. Therefore, TIMP-1 may be implicated in the pathogenesis of EF, which would be a good serological marker for disease activity, similar to gammaglobulins.
In summary, a deep wedge biopsy of the skeletal muscle, including the fascia, should be performed for diagnosis. In addition, MRI scan may be helpful in diagnosing atypical EF cases, when biopsy is unavailable or when the result of biopsy is doubtful; it can also readily monitor disease activity and therapeutic effect. Initial high dose steroid treatment is crucial in decreasing disease activity and duration.
 XML Download
XML Download