

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Wells' syndrome (WS), which was later named eosinophilic cellulitis, is a rare inflammatory dermatosis that was first described as a recurrent granulomatous dermatitis with concomitant eosinophilia by Wells in 19711. Although the pathophysiology of WS remains unclear, it is now believed to represent a specific hypersensitivity reaction to various exogenous and endogenous stimuli, such as insect bites, infections, drug eruption or underlying internal disorders2. Conversely, Churg-Strauss syndrome (CSS) is a multisystem granulomatous vasculitis that primarily affects patients with asthma. Classically, CSS is characterized by interstitial eosinophilic infiltrates, extravascular granulomas, which occur in a variety of organs, and peripheral eosinophilia. To date, only four cases of overlap between these two diseases have been described2-5. Here, we report a 57-year-old female with known CSS, who later developed a bullous skin eruption that was clinically and histologically consistent with WS.
CASE REPORT
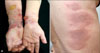
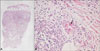
A 57-year-old woman came to our clinic with multiple erythematous tense vesicles and blisters with secondary yellowish crusts on both forearms, as well as multiple pruritic, erythematous, and annular plaques on her trunk (Fig. 1). She reported a history of asthma, bilateral maxillary sinusitis, vasculitis, and peripheral sensory-motor polyneuropathy in the lower extremities, all of which fulfilled the American College of Rheumatology criteria for CSS6. A skin biopsy from a blister on her arm revealed the presence of multiple intraepidermal vesicles and subepidermal blisters, with perivenular and interstitial infiltrations of lymphocytes and eosinophils in the superficial and deep dermis. Notably, these infiltrations also formed characteristic "flame figure" aggregates in the dermis, which was consistent with a diagnosis of WS (Fig. 2). Subsequent direct immunofluorescence microscopy did not show any immunoglobin or complement deposits in the lesional skin. An additional laboratory analysis revealed a white blood cell count of 5.71×109/L with peripheral eosinophilia (24.3%), an erythrocyte sedimentation rate of 112 mm/h, a C-reactive protein level of 4.22 mg/dl, and a negative anti-neutrophil cytoplasmic antibody (anti-MPO, p-ANCA). In the chest X-ray and chest computed tomography (CT), a diffusely increased opacity was noted in both lower lobes, whereas the echocardiography and abdomen CT did not indicate any associated pathologies. The patient was subsequently treated with oral predinosolone (1 mg/kg per day), which resolved the bullous skin lesions, as well as reduced the pain and pruritus for several days. The oral prednisolone was subsequently tapered over three weeks, and, 1 week after discontinuing all previous medications, she redeveloped the same rash. Consequently, treatment with oral predinosolone was restarted (1 mg/kg per day), and after a 1-month tapering, the skin lesions improved significantly. She is currently under regular surveillance.
DISCUSSION
WS is a rare, often recurrent, inflammatory disorder that is characterized by indurated urticarial erythematous plaques, similar to those seen in cellulitis. Pathologically, the characteristic feature of WS is "flame figures", which are histiocyte and giant cell aggregates surrounding collagen bundles that are coated with eosinophilic granules. These flame figures represent histological cutaneous reaction patterns specific to eosinophil-mediated dermatoses, most commonly WS, bullous pemphigoid, herpes gestationis, prurigo, eczema, or dermatophyte infections1. WS can present either solitary or multiple cutaneous lesions, which typically resolve spontaneously after several weeks with some residual atrophy and hyperpigmentation7. The other clinical manifestations of WS are quite diverse morphologically and range from cellulitis-like manifestations to less common bullous or vesicular lesions8. Affected patients often develop recurrences months to years after the initial presentation9, and peripheral eosinophilia is present in about 50% of all cases2.
CSS is characterized by the constellation of severe asthma, fever, hypereosinophilia, and symptoms of vascular impairment in various organ systems10. Although the pathogenesis of CSS remains unknown, it is now classified as a form of ANCA-associated small-vessel vasculitis (even though ANCA is present in less 50% of patients)11, and most current data suggests that CSS is induced by some allergenic or environmental trigger in pre-sensitized patients with asthma. Since CSS is primarily a clinical diagnosis, the American College of Rheumatology has established six separate criteria to facilitate the identification of affected individuals: (1) asthma (defined as a history of wheezing or diffuse, high-pitched wheezes upon expiration), (2) eosinophilia of >10% in differential white blood cell counts, (3) mononeuropathy (including multiplex) or polyneuropathy, (4) migratory or transient pulmonary opacities detected radiographically, (5) the presence of paranasal sinus abnormalities and (6) histologic evidence of perivascular eosinophil accumulation. In individuals with documented vasculitis, the presence of four or more of these criteria yields a diagnostic sensitivity and specificity for CSS of 85% and 99.7%, respectively. In the case of our patient, all criteria were fulfilled. Notably, the involvement of skin in CSS is not rare, occurring in roughly 40~70% of all patients, and classically presents as erythematous maculopapules, haemorrhagic lesions, or cutaneous and subcutaneous nodules2, while bullous lesions are substantially less common. Histologically, CSS is characterized by a triad of systemic vasculitis, eosoniphilic tissue infiltration, and extravascular granulomas. "Flame figures", however, have never been described in CSS, except in case reports of CSS-related WS. Thus, in our case, the bullous lesions were considered to be a WS-related symptom. Bullae are frequently present in WS5, and in most cases, they develop on top of urticarial erythematous plaques and cap a small area of the plataeu.
Hypothetically, CSS could induce WS via the pathogenic affects of the associated eosinophils that infiltrate through the skin. It is assumed that the eosinophilic inflammatory processes that target the skin, nerves, and blood vessels result from the underlying CSS pathophysiology. Accordingly, when diverted into the skin, which is possibly due to an additional unknown trigger, these same processes may produce "flame figures" histologically, and the bullous form of WS clinically4. Interestingly, Churg and Strauss suggested in their initial report that WS may be related to CSS, due to the similarity of the histopathologic changes associated with both conditions10. In regards to the treatment of CSS with WS, we found that prednisolone at a dose of 1 mg/kg was an effective therapeutic regimen5.
Notably, in all reported cases of CSS/WS overlap, CSS always preceded WS, suggesting that WS should be considered in the differential diagnosis of patients with CSS whose clinical presentation includes erythematous urticarial plaques. Although the association between these two conditions may be purely coincidental, it could also represent a common pathogenetic background, since both WS and CSS share many main features, including peripheral and/or tissue eosinophilia and widespread eosinophilic infiltration into multiple organs.
 XML Download
XML Download