

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Mal de Meleda (MDM) is an uncommon, autosomal recessive form of palmoplantar keratoderma (PPK), with most cases reported in descendants of the inhabitants of the island of Meleda (now Mljet), in Croatia1,2. Recently, a number of sporadic cases have also been reported in more widespread areas, such as the Middle East, Northern Africa, Turkey, Sweden and Taiwan3. We herein present a sporadic case of MDM in a 15-year-old Korean female who had several clinical obligatory features and mutations in the ARS gene, encoding SLURP-1. Interestingly, Korea is not only geographically the farthermost area from the endemic area, but is also traditionally a homogeneous society.
CASE REPORT
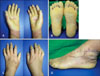
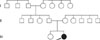
A 15-year-old female presented with diffuse palmoplantar thickening that had been present from birth. In the beginning, the lesions were limited to only her fingertips. However, with age, they progressively spread to the dorsal surfaces of the hands and feet. Since the age of ten, old, waxy ivory-yellow, palmoplantar hyperkeratotic plaques, with well-demarcated margins, accompanied with nail change, have been present. On cutaneous examination, we observed extensive diffuse hyperkeratosis of the palms and soles, which extended to the dorsal surfaces of the hands and feet (Fig. 1). She had knuckle pads on several interphalangeal joints and on both elbows. Conical tapering of the fingers and nail changes included thickening of the nail and hypercurvature. She also had a thin tongue, with several furrows at the distal portions of the tongue. The palmoplantar skin revealed an unpleasant smell, and maceration with hyperhidrosis, especially during the summer months. Teeth, hair, and general physical examination were unremarkable. None of the family members were affected and no consanguineous relationship was found (Fig. 2). The potassium hydroxide (KOH) mounts from the hyperkeratotic lesions of the palm, sole, dorsa of hands and feet, fingernail, and toenail, were all positive. Other laboratory work up was normal. Histopathologic findings of the sole, as well as the dorsa of the hand and foot, showed marked hyperkeratosis, acanthosis, and normogranulosis, without epidermolysis (Fig. 3A). PAS-positive spores and hyphae were present in the stratum corneum (Fig. 3B). Her genetic study detected mutations in SLURP-1 (Table 1). From these findings, she was diagnosed as MDM. She was topically treated with a mixture of 50% salicylic acid and steroid ointments, and systemically with oral antifungal and antibiotic agents. After combined treatments, the hyperkeratotic plaques showed some improvement, including reduction in thickness, especially in the erythematous dorsal borders, and the malodorous smell disappeared.
DISCUSSION
The PPKs are a heterogenous group of disorders, characterized by thickening of the skin of the palms and soles4. They can initially be categorized, based on whether they are inherited or acquired. Then they can be further subdivided, based on clinical patterns. MDM, one of the autosomal recessive forms of PPK, is clinically characterized by a well-demarcated erythema and hyperkeratosis of the palms and soles, that usually occurs soon after birth. The hyperkeratosis spreads slowly to the dorsal aspects of the hands and feet, which is referred to as transgradiens. Many patients will experience bothersome pain due to fissures. Hyperhidrosis with maceration is also characteristic of MDM, and is often accompanied by malodor. In addition, patients may develop keratotic plaques over joints, nail abnormalities, brachydactyly, pseudoanihum and perioral erythema5.
Our case showed the following compatible clinical features: "glove-and-socks" distribution of the keratoderma, with a sharp demarcation that appeared after birth, and progressively extended to the dorsa of the hands and feet; nail changes; conical tapering of the fingertips; and malodorous palmoplantar hyperhidrosis.
In 2001, mutations in the gene encoding SLURP-1 located on the chromosome 8q24.3 were found as the cause of MDM6. It is currently identified that SLURP-1 is an epidermal secreted neuromodulator that influences both epidermal homeostasis and inhibition of tumor necrosis factor-alpha release by macrophage, during the wound healing process. Such roles would explain the hyperproliferative and inflammatory clinical characteristics of the MDM7. Herein, we analyzed our patient for mutations in SLURP-1. Mutations analysis revealed compound heterozygous mutation in exon 3 of the ARS gene. A heterozygous G to A transition at neucleotide 256 resulted in a missense mutation in SLURP-1 at the amino acid position 86. This missense mutation has been previously reported in patients from Palestine and Taiwan1,2. She also exhibited a heterozygous C to T transition at nucleotide 286, leading to conversion of the arginine risidue to a stop codon at amino acid 96. This nonsense mutation has been described in patients from Turkey and Croatia1.
Although MDM is not a life-threatening disease, its chronic disfigured hyperkeratotic lesions with malodor can lead to impaired social activity and occupational hazard. Treatment is extremely important at this point, even though it is not yet possible to completely heal this genetic disorder. Therapeutic options include urea containing moisturizers or keratolytics, such as salicylic acid. The aromatic retinoids etretinate and acitretin usually produce improvement of PPK8. Of importance, however, is that early and long-term use of retinoid associates with several well-known adverse effects. In addition, antibiotic and antifungal therapy is helpful, if needed9. Although it is not a pathognomonic finding, many patients with MDM have an increased risk of either bacterial or fungal infection that may increase the skin thickening and may result in an unpleasant smell. Our patient has taken antibiotic and antifungal treatments, resulting in not only disappearance of malodor, but also thinning of the dorsal hyperkeratotic plaques. Because the various treatments may offer symptomatic relief, it is important to be aware that continuous treatment is necessary in the management of this life-long disease.
MDM was originally described in patients from the island of Meleda in Croatia. However, recently, MDM has been observed in more widespread countries beyond the Mljet island, and several sporadic cases have been reported3. Our case has an important meaning, in that it has occurred in Korea, currently the farthest country from the Mljet Island and with no connection in history. In addition, we emphasize that a comprehensive long-term, follow-up plan, particularly if supported by effective secondary infection management strategies, can be surprisingly helpful.


 XML Download
XML Download