

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Liposarcomas are deep-seated soft tissue tumors in adults that originate in primitive mesenchymal cells1. Liposarcomas represent 15% to 20% of all soft tissue sarcomas and the annual incidence of all types of soft tissue liposarcoma is estimated at 2.5 per million. Liposarcoma is the most common sarcoma of deep soft tissues in adults, and the average age at presentation is in the 50s. The common sites of liposarcoma are the extremities, particularly the thigh, buttocks or retroperitoneum1,2. Although liposarcoma can also extend to subcutaneous tissue from a fascial plane, they rarely occur in the dermis and upper subcutaneous layer3. Liposarcomas for practical purposes do not generate from lipomas. The diagnosis of liposarcoma is made based on the presence of lipoblasts that are immature fat cells characterized by a hyperchromatic nucleus indented or scalloped by cytoplasic fat vacuoles. We describe here a unique case of subcutaneous myxoid and round cell liposarcoma arising in the left flank with a literature review.
CASE REPORT
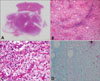
A 28-year-old man presented at our clinic with a rapid growing mass located on his left flank. The patient had first recognized tensile force on the area and then found a soft, painful mass two months prior to presentation. There was no episode such as trauma or physical injury before development of the mass. Upon physical examination, we noticed a subcutanous mass that was tender, mobile and soft with a diameter of about 3 cm (Fig. 1A). The differential diagnosis included lipoma, angiolipoma, leiomyoma and neurilemmoma. No abnormalities were detected upon laboratory work up. Computed tomography (CT) revealed a well-defined, approximately 2.5×2.3 cm sized soft tissue density mass in the subcutaneous area on the left flank without significant lymphadenopathy (Fig. 1B). Histopathologic examination showed a relatively well circumscribed, nonencapsulated tumor immediately under the dermis with an infiltration of the lateral margin (Fig. 2A). The tumor consisted of dispersed atypical lipoblasts and plentiful, plexiform "chicken-wire" capillary vasculatures in a prominent myxoid stroma (Fig. 2B). Some areas showed collection of the uniform round to oval shaped primitive nonlipogenic mesenchymal cells. Atypical proliferating lipoblasts showed varying degrees of differentiation and small signet-ring lipoblasts were occasionally seen (Fig. 2C). On immunohistochemistry, alcian blue stain demonstrated mucin deposits in the surrounding stroma with mucopolysaccharide lakes (Fig. 2D), but CD34 and S100 were negative. The tumor was eventually diagnosed as a myxoid and round cell liposarcoma. The patient was then transferred to the department of plastic surgery and the lesion was widely excised with a flap. There was no recurrence within seven months of the excision; however, long-term follow-up was recommended.
DISCUSSION
Liposarcoma is diagnosed when there is convincing evidence of the synthesis and storage of fat by tumor cells, the hallmark of which are the lipoblasts1. Lipoblasts are immature fat cells characterized by a hyperchromatic nucleus that is indented or scalloped by cytoplasmic fat vacuoles. Other histopathological appearances are highly variable and depend on the particular subtype. Immunohistochemistry is of limited value in the diagnosis of liposarcoma, although it may help to reveal the identities of various sarcomas such as malignant schwannoma, rhabdomyosarcoma, and leiomyosarcoma. The pathological classification of liposarcomas have undergone change. In 1994, five histopathological subtypes were recognized as well-differentiated, myxoid, round cell, dedifferentiated and pleomorphic variants by the World Health Organization (WHO)2. Myxoid liposarcoma resembles immature fat and small, uniformly bland spindle cells set in a myxoid matrix with plexiform vessels. Round cell liposarcoma is characterized by sheets of primitive round cell and commonly coexist with myxoid variants. Even if still classified by the WHO as two distinct subtypes, myxoid and round cell liposarcoma share both clinical and morphologic features, and lesions combining both patterns are frequent. In 2004, newer classification of liposarcomas was made, which divided them into three biologic types encompassing five subtypes based on strict morphologic features, natural history and cytogenic aberrations: (1) myxoid/round cell, (2) well-differentiated/dedifferentiated, and (3) pleomorphic4,5. Both myxoid and round cell liposarcomas show a specific, reciprocal, chromosomal translocation, most commonly t(12;16)(q13;p1), which results in rearrangement of the CHOP gene6.
Myxoid and round cell liposarcoma account for about 30~35% of all liposarcomas. The tumors generally affect young to middle-aged adults with a peak in the fourth to fifth decade and a slight male predominance2. The clinical manifestation of this variant presents as a large, painless mass located in deep soft tissue and involves the thigh in over two-thirds of cases. The tumor is described as dome-shaped or polypoid, and measures between 1 and 19.5 cm7. In cases of typical myxoid and round cell liposarcoma at deeper soft tissue, it is necessary to distinguish the tumor from intramuscular and juxta-articular myxoma, aggressive angiomyxoma, myxofibrosarcoma and myxoid chondrosarcoma8. Interestingly, our case differed from previous reports in that it was detected on the upper subcutis of the flank in a relatively young male and accompanied with pain. Indeed, its clinical presumptive diagnosis was a subcutaneous benign mass such as a lipoma, angiolipoma, leiomyoma or neurilemmoma. Lipoma is the most common soft tissue tumor with a predilection for the trunk and proximal limbs. Morphologically, angiolipoma is similar to lipoma, but intermittently painful. These characteristics could explain why we were unable to identify what the tumor was before the histology was obtained from the biopsy.
Liposarcoma are on a morphological continuum in which tumor progression from low-grade myxoid to high grade hypercellular or round cell areas may be observed. Wide agreement exists in considering round cell liposarcoma as a poorly differentiated myxoid liposarcoma4. Purely myxoid lesions are characterized by a 5-year survival rate of approximately 70%, which drops to approximately 20% in the case of round cell tumors. Two independent studies have demonstrated that the presence of a round cell component ranging between 5 and 25% is associated with a significant worsening of prognosis when compared to pure myxoid lesions7,9. In addition, patients who present with multifocal disease have a poor prognosis and the presence of necrosis and p53 overexpression have been found to be independent predictors of poor prognosis10. There is no significant relationship between tumor size and clinical outcome. Local recurrence has been reported with an unusually high incidence of soft tissue and bone metastases to the lung. However, in 1998, Dei Tos et al.3 reported their personal experience that liposarcoma occurs primarily in the skin, and that despite an apparent tendency to show high-grade morphological features, it appears to behave in a relatively benign fashion, in stark contrast to their deep-seated counterparts.
Surgery remains the mainstay of therapy for liposarcoma Vol. 23, No. 3, 2011 341 in the control of local disease. Excision needs to be carefully planned using CT or magnetic resonance imaging11. Liposarcoma can often be managed if surgical treatment is carefully planned and adequate clean margins are achieved. Nevertheless, patients with round cell and pleomorphic liposarcoma will develop distant metastasis, usually to the lungs, bone or fat pad sites in the retroperitoneum and trunk. Since the high rate of response of myxoid/round cell liposarcoma to ifosfamide-based chemotherapy has been observed in the metastatic setting, neoadjuvant or adjuvant chemotherapy for high-risk primary liposarcoma may help decrease the frequency of distant metastases, and thus increase overall survival. To lower the recurrence or distant metastasis rates, radiotherapy is also recommended before or after surgery12.
Liposarcoma is rarely reported in the Korean dermatologic literature, and only five cases of liposarcoma have been described since 199613-16. All of these cases involved the extremities, especially the thigh, and with the exception of one case in the dermis, all of these cases occurred in the deep soft tissue. If liposarcoma is detected in the trunk of young adults, it can be easily mistaken for a benign tumor such as a lipoma or angiolipoma. Herein, we report a case of myxoid and round cell liposarcoma affecting the subcutis of the left flank. To avoid missing the optimal timing for treatment, clinicians should be aware of the possibility of malignant diseases, especially when rapid growth of the subcutaneous tumor is observed.

 XML Download
XML Download