

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Atrichia with papular lesions (APL) is a rare form of irreversible complete loss of hair on the scalp and entire body, and is known to be inherited in an autosomal recessive manner1-4. Clinically, APL is characterized by total hair loss, including scalp, face, axilla, and body hair, during the first few months of life. This is followed by the appearance of multiple follicular papules that consist histopathologically of keratin-filled cysts and are especially prevalent on the face and extensor surfaces of the extremities.
Recently, molecular analysis has been used to diagnose a patient with APL, and studies of numerous APL families have implicated mutations of the hairless (HR) gene in the pathogenesis of this disorder4-6. Several mutations that map to chromosome 8p12 have been identified, and increasing numbers of HR gene mutations are being reported.
Most APL patients have homozygous HR gene mutations, and have been discovered in consanguineous families living within a restricted geographical region, implying that these mutations became fixed owing to a high degree of consanguinity1-3. However, the number of sporadic cases occurring in offspring of unrelated parents has been increasing7-11. Sporadic patients with non-consanguineous parents have been misdiagnosed with alopecia universalis (AU) frequently, as APL and AU are clinically very similar. After unsuccessful treatment for AU, they were subsequently diagnosed as APL by molecular analysis5,12. Thus, the identification and evaluation of HR gene mutations in patients with total hair loss are important for differentiating between APL and AU.
In this study, the HR genes of AU patients who showed little or no response to treatments for restoring hair growth were examined and compared with the HR genes of normal healthy controls, and a novel missense mutation of the HR gene was identified in a Korean boy who finally diagnosed with APL.
MATERIALS AND METHODS
Human subjects
The study examined 11 patients presumed to have AU who were seen in the dermatology clinic at Busan Paik Hospital, Busan, Korea and had little or no response to systemic and topical hair restoration treatments. The molecular analyses of HR genes in the AU patients and 50 normal healthy controls were compared. Among the 11 patients with presumed AU, one male was diagnosed with APL based on his HR gene analysis. Subsequently, his immediate family and several close relatives were also included in this study. He had no family history of APL and no history of parental consanguinity.
The proband was a 4-year-old boy who was born with nearly normal scalp hair but sparse eyebrows and eyelashes. The scalp hair began to shed soon after birth and never regrew. He progressed to complete and permanent hairlessness by the age of 3 years. He did not respond to standard systemic and topical immunotherapy for AU, which included potent steroids and diphencyprone (DPCP). Multiple follicular papules began to appear on his face, chest, back, elbows, and knees when he was 3 years old, and the eruptions gradually increased in number. His nails, teeth, and sweating were normal, and there was no evidence of ectodermal dysplasia.
Clinical material
We obtained blood samples from 11 AU patients (including proband), family members of the proband, and 50 normal healthy controls. To confirm APL diagnosis, punch biopsies from the right occipital region of the scalp and the left thigh, and a follicular papule from the left knee of the proband were obtained under local anesthesia. The biopsy samples were fixed in 4% paraformaldehyde, embedded in paraffin, cut into 4-µm-thick sections, and stained with hematoxylin and eosin using standard protocols.
Mutation detection
To detect mutations in the human HR gene, all exons and splice junctions of the HR gene from each subject's genomic DNA were amplified by the polymerase chain reaction (PCR), purified using GeneAll® Expin™ Gel SV (GeneAll Biotechnology, Seoul, Korea), and sequenced directly in an ABI Prism 3100 automated sequencer, using a ABI Prism Dye Terminator Cycle Sequencing Ready Reaction kit (SolGent, Daejeon, Korea). The PCR conditions have been described elsewhere5. The mutation was identified by comparisons of the HR sequences among the AU patients and control subjects, using the ClustalX multiple sequence alignment program (Strasburg, France). The mutation was verified in the patient by comparing the BseRI (New England Biolabs, Beverly, MA, USA) restriction pattern of his HR gene with those of several family members (his parents, maternal and paternal grandparents, an aunt, two uncles, and a brother), the AU patients, and the control individuals.
RESULTS
Clinical findings consistent with atrichia with papular lesions
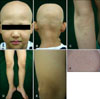
At the time of examination, the proband had only two mature black hairs remaining, in the right occipital region. We also observed absence of body hair, and sparse eyebrows and eyelashes (Fig. 1A, B). More than 100 small papules were present over the scalp, face, chest, back, both elbows, and both knees (Fig. 1A, C, D). There were also several fine, whitish, hypopigmented streaks on the scalp. On careful examination of the patient, a novel clinical sign that has not been described in previous reports of APL was observed, namely, hypopigmented macules on the entire body, especially on both thighs (Fig. 1E, F). The patient had no abnormalities of the nails, teeth, sweating, or laboratory findings, including vitamin D metabolites. He also has no major developmental or growth deficiencies.
Histopathology
1) Scalp
Only two mature black hairs were present in the right occipital region of the proband. The skin biopsy specimen from the scalp showed a single intact mature hair and a markedly reduced number of hair follicles. A few vellus hairs and some granulomatous changes were observed around an anagen hair follicle. There was mild perivascular lymphocytic infiltration limited to the infundibulum and isthmus level with no inflammation around the hair bulb (Fig. 2A).
2) Thigh
The skin biopsy specimen of multiple tiny whitish macules on the thigh showed the loss of hair follicles with remnant arrector pili muscles. There was no remarkable change in pigmentation, including melanin and melanocytes, in the basal layer of the epidermis (Fig. 2B).
3) Knee
A biopsy of a follicular papule on the knee showed a keratinous cyst containing fine laminated eosinophilic keratinous material. The cyst wall consisted of several layers of keratinocytes showing gradual flattening, and a granular layer was present in the cystic epithelium. The histopathological findings of the papule on the knee were compatible with an epidermal inclusion cyst (Fig. 2C).
Identification of a mutation in the human HR gene
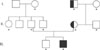
The proband showed a novel G-to-A transition at nucleotide position 191 in exon 5, resulting in the replacement of a glycine residue by a glutamate; this was called a G64E missense mutation (Fig. 3). The G64E mutation eliminated a restriction site for the endonuclease BseRI and was used as a screening assay in 9 relatives, 10 other AU patients, and 50 Korean control individuals. The proband's mother and maternal grandfather were heterozygous carriers of the G64E mutation (Fig. 4). But, the chromosomes containing the HR gene in all of the other AU patients and healthy controls were wild type. Therefore, this mutation was not a polymorphism. The absence of this mutation in the control chromosomes and the nature and severity of the clinical findings indicate that this was a pathogenic mutation in this patient.
DISCUSSION
Relatively little is known about the mechanism of APL. Although hair follicle development appears to be normal in APL, a defect in the follicular cycling system must induce hair loss because normal hair is present at birth but is subsequently lost during the first few months of life. Panteleyev et al.13 postulated that hair regrowth in APL is disrupted because the lower follicle disintegrates during catagen and the normal juxtaposition of epithelial stem cells in the bulge and mesenchymal cells in the dermal papilla of the hair follicle is not re-established during telogen14. This interferes with mesenchymal-epithelial interactions, which are considered to be necessary for re-entry into anagen, and atrichia is the result14. Consequently, the architecture of hair follicles with short catagen stages, including those of vibrissae, eyelashes, and eyebrows, may not be disrupted sufficiently during catagen, and these hairs may be preserved in APL patients15. Recent studies have suggested that HR protein acts as a nuclear receptor co-repressor in concert with the vitamin D receptor gene and represses the expression of WISE protein, which inhibits Wnt signaling16,17. The follicular cysts seen in APL patients may arise from follicular remnants that survive after the follicle breaks apart during catagen13,18. As the cyst epithelium expresses keratin K17, which normally occurs in the outer root sheath of hair follicles, cysts in APL patients appear to arise from the bulge of the hair follicle13,19. The regression of these follicular papules in older patients has been reported, although the mechanism is unclear4.
Approximately 34 mutations of the HR gene, including the one discovered in this study, have been reported6,10-12,20-22. These include missense, nonsense, deletion, insertion, splice-site, and compound heterozygous mutations10,11,23-25. This study reports a novel G-to-A transition at nucleotide 191 in exon 5, resulting in the replacement of a glycine residue by a glutamate residue. This new missense mutation in the HR gene is called the G64E mutation. This particular mutation has not been reported previously, and this is only the second case report of APL occurring in a Korean family.
Although different mutations of the HR gene have been reported, the clinical manifestations of APL exhibit little variation, and most APL patients have the same degree of clinical severity. There do not appear to be any particular "hot spots" or common locations for mutation in the HR gene or any genotype-phenotype connections in APL patients. Also, there is no information on HR protein levels in affected patients, particularly the actual amount of altered HR protein expressed in the skin. Therefore, to diagnose APL, it is important to sequence all coding regions, including exon-intron junctions, of the entire HR gene.
APL is usually inherited in an autosomal recessive manner. Most reported cases of APL have been discovered in consanguineous families living within a restricted geographical region. However, there are some reports of APL patients with compound heterozygous mutations in small non-consanguineous families7-11. In present study, the novel missense mutation was not inherited as an autosomal recessive, as the patient's mother and maternal grandfather were heterozygous carriers of the G64E mutation, whereas his father had the wild-type HR gene. There are three possible explanations for proband's homozygosity: 1) Proband's father is not his biological father. 2) The father has a large heterozygous deletion in the same region which cannot be recognized by sequencing and which was inherited by the proband. 3) The proband had a spontaneous mutation of the same type as his mother's (G-to-A transition at position 191 in exon 5), which seems to be less likely. It seems that isolated cases of APL may be more common than previously thought, because of a lack of awareness and the misdiagnosis of APL as AU or vitamin-D-dependent rickets, which may present as atrichia in the first few months of life and as follicular cysts in the first few years of life11,17.
On careful examination of this patient, hypopigmented macules and streaks were observed. Hypopigmented streaks on the scalp in APL patients have been reported before6. However, follicular hypopigmented macules over the entire body, especially on both thighs, have not been reported, and may be novel clinical signs of APL. The mechanism and pathogenesis of the hypopigmented streaks or macules in APL is unknown, but it may be that the HR gene mutation could play a role in the development of the follicular hypopigmented macules.
In summary, we report a new case of APL in a nonconsanguineous family that was diagnosed based on clinical findings and molecular data and involved a novel G-to-A transition at nucleotide 191 in exon 5. In addition, a novel clinical sign of APL was observed, namely, follicular hypopigmented macules on the entire body, especially on both thighs. This particular mutation has not been reported previously, and this is only the second case report of APL occurring in a Korean family. These data extend our knowledge of mutations in the HR gene, and suggest that children with presumed AU in non-consanguineous families that do not respond to standard treatment modalities may warrant testing for HR gene mutations to avoid unnecessary treatments.


 XML Download
XML Download