

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Human epidermal growth factor receptor (EGFR) is dysregulated in many solid tumors, making it an attractive target for anticancer therapy. Indeed, monoclonal antibodies, such as cetuximab that block EGFR, and small molecules, such as gefitinib and erlotinib that inhibit the tyrosine kinase activity of this receptor, are used for the treatment of solid tumors, including lung cancer and colorectal cancer1-5.
EGFR is primarily expressed in undifferentiated, proliferating epidermal keratinocytes6. It is also expressed in sebaceous glands, the outer root sheaths of hair follicles, and the capillary system7,8. The activation of EGFR by its ligands, including transforming growth factor alpha (TGF-α) and heparin-binding epidermal growth factor (HB-EGF) in the skin, has been shown to regulate normal keratinocyte proliferation, differentiation, migration and survival via downstream signal transduction cascades such as the mitogen-activated protein kinase (MAPK) pathway, the phosphatidylinositol 3 kinase pathway and the signal transducer and activator of transcription pathway3,6,9,10.
EGFR inhibitors induce skin rashes, such as acneiform eruptions, by blocking EGFR in the normal skin of the scalp, face, and upper chest, where high levels of EGFR expression and a high density distribution of sebaceous glands are present1,2,4,5,11. Inhibition of EGFR signaling in basal keratinocytes leads to their immediate growth and migratory abnormalities along with inflammatory changes6. In addition, the blockade of EGFR induces derangement of chemokine expression in keratinocytes, leading to enhanced skin inflammation12. These facts demonstrate that the blockade of EGFR in keratinocytes may be responsible for the acneiform eruptions that are induced by EGFR inhibitors.
Sebaceous glands, which are contiguous with the basal layer of the skin, show high EGFR expression, and play important roles in inflammatory acne, producing sebum and inflammatory biomarkers. They may also be involved in the pathogenesis of EGFR inhibitor-induced acneiform eruptions. Therefore, we studied the effects of an EGFR inhibitor (cetuximab) and of EGFR ligands, such as epidermal growth factor (EGF) and TGF-α, on the expression in cultured sebocytes of inflammatory biomarkers. We used reverse transcription-polymerase chain reaction (RT-PCR), immunocytofluorescence (ICF) and Western blot assays to investigate the role of sebaceous glands in EGFR inhibitor-induced acneiform eruptions.
MATERIALS AND METHODS
Materials
1) Specimens
Specimens for sebocyte cultures were obtained from the occipital scalp region of patients with male pattern hair loss during hair transplantation. Informed consent was obtained from each patient.
2) Culture medium
Either Dulbecco's modified Eagle's medium (DMEM; Gibco BRL, Grand Island, NY, USA) supplemented with penicillin (100 U/ml), streptomycin (100 µg/ml) and 20% heat inactivated fetal calf serum (Hyclone, Waltham, MA, USA), or keratinocyte growth medium (KGM; Gibco BRL) supplemented with penicillin (100 U/ml), streptomycin (100 µg/ml) and fungizone (250 µg/ml) were used as media.
3) EGFR inhibitor, EGF and TGF-α
Erbitux®, an EGFR inhibitor was purchased from ImClone/Bristol-Myers Squibb (New York, NY, USA). EGF and TGF-α were obtained from Millipore Corporation (Billerica,
MA, USA).
4) Antibodies
Primary antibodies used were interleukin (IL)-1 (R&D Systems, Minneapolis, NM, USA, 1:100), IL-6 (Chemicon, Billerica, MA, USA, 1:100), tumor necrosis factor-α (TNF-α) (Chemicon, 1:100), peroxisome proliferator-activated receptor-γ (PPAR-γ) (Cell Signaling, Danvers, MA, USA, 1:50), and EGFR (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Fluorescein isothiocyanate (FITC) conjugated anti-rabbit antibodies (Pierce, Rockford, IL, USA, 1:60) and FITC conjugated anti-mouse antibodies (DAKO, Produktionsvej, Denmark, 1:60) were used as secondary antibodies.
Methods
1) Human sebocyte culture
Primary cultures of sebocytes were maintained according to the method described previously13. Sebaceous glands were isolated from dissected hair follicles under a binocular microscope and transferred to tissue culture dishes. Cells were maintained in DMEM at 37℃ in a humidified 5% CO2 atmosphere. The explants were left for 5 days. Then, the medium was changed to KGM media and was changed every 3 days. After cell outgrowths became subconfluent (70% of the culture dish), cells were harvested with 0.25% trypsin and 10 mM ethylenediamine tetraacetic acid (EDTA) in Hank's balanced salt solution and subcultured with a split ratio of 1:3. The cells obtained after the second passage were used in our study.
2) Addition of EGFR inhibitor and EGFR ligands into sebocyte cultures
Cultured sebocytes were divided into four groups. One group, a control, was not treated. The other groups were treated with 10 ng/ml of EGF, or 10 ng/ml of an EGFR inhibtor, or 5 ng/ml of TGF-α.
3) RT-PCR
(1) Total RNA extraction
Total RNA was isolated from cultured cells using TRIzol reagent (Gibco-BRL, Grand Island, NY, USA) according to a modified acid phenol method14.
(2) Isolation of messenger RNA (mRNA)
mRNA was isolated from total RNA using Dynabead mRNA purification kits (Dynal, Lake Success, NY, USA).
(3) RNA amplification
cDNA was synthesized from 5 µg of total RNA using cDNA synthesis kits containing ImPromIITM reverse transcriptase and random primers (Promega). PCR amplification was done using GoTaqFlexi DNA Polymerase. All amplifications (except IL-1) were done for 35 cycles under the following conditions: 95℃ for 1 minute, 56℃ for 1 minute, and 72℃ for 1 minute. IL-1 amplifications were done for 25 cycles using the same time/temperature parameters.
(4) Semiquantitatve RT-PCR analysis
To assess mRNA expression levels of IL-1, IL-6, TNF-α, PPAR-γ and EGFR, semiquantitative RT-PCR analyses were done using microcomputer imaging device Image J software (National Institutes of Health, Bethesda, MD, USA). Expression of mRNA for β-actin was used as a control marker. Differences in the expression of inflammatory biomarkers between treated and untreated (control) groups were expressed as the ratio of the treated group to the control group.
4) Immunocytofluorescence
ICF was measured for cells fixed to a four-well chamber slide with 4% paraformaldehyde at room temperature for 15 minutes. After washing with phosphate buffered saline (PBS) for 5 minutes (three times), slides were preincubated for 1 hour in 10% bovine serum albumin (in PBS) to prevent non-specific antibody binding. Next, the slides were incubated overnight at 4℃ with primary antibodies . Primary antibodies were those raised against IL-1, IL-6, TNF-α, PPAR-γ and EGFR. Non-specifically bound primary antibody was removed by four sequential washes in PBS (5 minutes each). After washing with PBS, slides were incubated in the presence of secondary antibody for 1 hour at room temperature. Analyses of intense immunoreactivity were performed using microcomputer imaging device Image J software.
5) Western blots
Cells were lysed in radioimmune precipitation assay buffer {150 mM NaCl, 50 mM Tris-HCl (pH 7.2), 1% deoxycholic acid, 1% Triton X-100, 0.25 mM EDTA (pH 8.0), 0.2% sodium fluoride, 0.1% sodium orthovanadate} supplemented with protease inhibitor cocktail. Equal amounts of total cell lysates were resolved by sodium dodecylsulfate-polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes, blotted with the appropriate antibodies, and analyzed using enhanced chemiluminescence and autoradiography. Relative intensity of the bands was measured using microcomputer imaging device Image J software.
RESULTS
Treatment-induced mRNA expression of proinflammatory biomarkers
All proinflammatory biomarkers, except for IL-1 in the EGF-treated group and IL-6 in the EGFR inhibitor-treated group, showed decreased mRNA expression in all of the treatment groups compared with the control group (Table 1). An increase in IL-1 expression was found in the EGF-treated group, and an increase in IL-6 expression was found in the EGFR inhibitor-treated group. A decrease in the expression of IL-1 was observed in the EGFR inhibitor-treated and TGF-α-treated groups, and a decrease in the expression of IL-6 was observed in the EGF-treated and TGF-α-treated groups. A decrease in the expression of TNF-α, PPAR-γ, and EGFR was shown in all of the treated groups. However, there were no significant differences in mRNA expression of inflammatory biomarkers between the treated groups and the control group (p>0.05) (Figs. 1, 2).
Treatment-induced expression of proinflammatory biomarkers using ICF
All proinflammatory biomarkers, except for IL-1 in the EGF-treated group and PPAR-γ and EGFR in the TGF-α-treated group, showed decreased expression in all treated groups compared with the control group (Table 1). An increase in the expression of IL-1 was found in the EGF-treated group (p<0.05), but other biomarkers in the same group showed decreases. We found decreases in the expression of IL-1, IL-6, TNF-α, PPAR-γ and EGFR in the EGFR inhibitor-treated group. Downregulation of IL-1, IL-6 and TNF-α, and upregulation of PPAR-γ and EGFR were shown in the TGF-α-treated group. Only the increase in IL-1 in the EGF-treated group was significantly different than the control group (p>0.05) (Fig. 3).
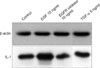
Effects of treatment on expression of IL-1 using Western blots
Expression of IL-1 in Western blots was increased in the EGF-treated group and decreased in the EGFR inhibitor-treated group when compared with the control group (Table 1). In contrast to an increase in the EGF-treated group and a decrease in the EGFR inhibitor-treated group, there was no change in the expression of IL-1 in the TGF-α-treated group (Figs. 4, 5).
DISCUSSION
EGFR is the cell surface receptor for members of the EGF family of extracellular protein ligands such as EGF, TGF-α, HB-EGF, amphiregulin and epiregulin3,6. Apart from direct activation by its protein ligands, EGFR can be activated by heterogeneous ligands or EGFR ligand-independent mechanisms including a variety of G-protein-coupled receptor ligands and by proinflammatory cytokines binding their specific receptors and integrins15-18.
Abnormal signaling through the EGFR pathway is associated with neoplastic development including cell proliferation, migration, stromal invasion, resistance to apoptosis, and angiogenesis5,19,20. The high frequency of abnormalities in EGFR signaling in human carcinomas has shown that the inhibition of EGFR can impair tumor growth, and has made EGFR a promising target for cancer treatment5,19,20. EGFR inhibitors targeting the EGFR pathway inhibit the proliferation of cancer cells through EGFR blockade-induced upregulation and/or accumulation of the cyclin-dependent kinase inhibitor p27Kip1and/or p21CIP1/WAF1, and the inhibition of the ERK1/2 MAPK pathway. EGFR inhibitors also inhibit angiogenesis and metastasis via downregulation of the proangiogenic factor and induce apoptosis through upregulation of proapoptotic molecules and downregulation of antiapoptotic molecules9,21.
Unfortunately, blocking the EGFR pathway with a monoclonal antibody such as cetuximab, or with small molecules that inhibit the tyrosine kinase activity of EGFR such as gefitinib and erlotinib, leads to various dermatological side effects in the scalp, face and upper chest, most frequently an acneiform eruption5,22. Although the overexpression of EGFR in the epidermis, sebaceous glands and hair follicles presents as a side effect on skin, hair and nails, the pathophysiology by which inhibition of EGFR leads to various dermatologic reactions remains largely unknown23,24. Data from a large number of clinical trials suggest that there is a direct relationship between rash incidence and response to EGFR inhibitors. Duvic proposed in his report that an acneiform eruption is a classic effect of EGFR inhibitors that is thought to be a direct result of EGFR blockade in the hair follicle1. Although data on mechanisms is limited, EGFR inhibitor-induced skin rash can reasonably be derived from impairment of multiple EGFR-dependent homeostatic functions of skin25.
The disruption in growth and differentiation of hair follicles by EGFR inhibitors may contribute to follicular acneiform eruption, neutrophilic folliculitis, and perifolliculitis3,5. This disruption may cause a mechanical rupture of the hair follicle, leading to hyperkeratosis, follicular plugging, and eventual inflammation of the pilosebaceous unit. It should be stressed that no comedones precede the follicular papules or pustules of an acneiform eruption5,11. Surguladze and colleagues showed that a monoclonal antibody targeting murine EGFR caused a neutrophil-rich hair follicle inflammation in mice26. The effect was preceded by the appearance of lipid-filled hair follicle distensions adjacent to enlarged sebaceous glands. In addition, the cytokine TNF-α, which is localized immunohistochemically to the affected region of the pilosebaceous unit, was specifically up-regulated by murine EGFR. Therefore, it is thought that blockade of the EGFR not only in keratinocytes but also in sebaceous gland cells may be responsible for EGFR inhibitor-induced skin rashes. EGFR inhibitors may also directly affect the immune system by reversing the suppression of chemokine production, resulting in leukocyte chemotaxis and infiltration of the skin5,11. There is evidence that focal keratinocyte necrosis due to persistent EGFR inhibition activates and sustains immune cell recruitment and leads to folliculitis. Cultured keratinocytes display a massive upregulation in the levels of chemokines such as CCL2, CCL5 and CXCL10, when EGFR is blocked15. As described above, the expression of TNF-α can be upregulated in EGFR inhibitor-induced acneiform eruption7. It initiates a program of increased expression of inflammatory mediators such as cytokines and chemokines6. The current study, which was performed to evaluate the effects of EGFR inhibitors on cultured sebocytes, showed that inflammatory biomarkers, such as IL-1, IL-6, TNF-α, PPAR-γ and EGFR, were not upregulated in cultured sebocytes treated with EGFR inhibitors (10 ng/ml Erbitux®). It has been demonstrated that EGFR activation is involved in the downregulation of chemokine expression in human keratinocytes12,13. In particular, EGFR activation by TGF-α can potently downregulate the levels of TNF-α- or interferon-gamma-induced RANTES and monocyte chemotactic protein-1, thus potentially opposing the attraction of neutrophils, monocytes/macrophages and dendritic cell precursors to skin lesions12. Migration of T cells could also be impaired due to EGFR-driven suppression of the CXC chemokine receptor 3 ligand IP-10. Although chemokine expression in cultured sebocytes treated with EGFR ligands, such as EGF and TGF-α, was not evaluated in our study, a decrease in the expression of inflammatory biomarkers including IL-1, IL-6, TNF-α, PPAR-γ and EGFR in the groups treated with 10 ng/ml EGF and 5 ng/ml TGF-α was observed; only IL-1 expression in the EGF-treated group showed an increase. Inflammatory biomarkers, which are present in normal sebaceous glands, are upregulated under stressful conditions such as the treatment with Propionibacterium acnes, lipopolysaccharide and substance P27.
In short, our study demonstrated that exposure to an EGFR inhibitor, which is often a cause of acneiform eruptions, decreases the expression of inflammatory biomarkers in cultured sebocytes, and that EGFR activation through EGFR ligands like EGF and TGF-α shows a decrease in the expression of inflammatory biomarkers in cultured sebocytes. However, statistically significant differences were not found. Further studies are still needed to (1) determine whether there is a correlation between abnormalities in sebaceous glands in vivo and the presence of EGFR inhibitors, and (2) to determine the mechanism of EGFR inhibitor-induced acneiform eruption.





 XML Download
XML Download