

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Mastocytosis is a rare disease characterized by a primary pathological increase in the number of mast cells. It may present with a variety of clinical signs and symptoms and the prognosis varies. The skin is the most commonly involved organ in all types of mastocytosis. Cutaneous mastocytosis (CM) is a heterogeneous disorder that is divided into three major variants: urticaria pigmentosa (UP), diffuse cutaneous mastocytosis (DCM), and mastocytoma1-3. Bullous eruption is most commonly associated with DCM, although bullae can occur in all forms of CM. It is important to differentiate DCM from other bullous skin disorders observed in infants, such as epidermolysis bullosa, bullous congenital ichthyosiform erythroderma, and staphylococcal scalded skin syndrome. Here, we report a case of DCM presenting with generalized bullae.
CASE REPORT
A 9-month-old female infant presented with generalized blisters that were first noted when she was three months old. The lesions were first observed on the hands and feet, and spread to the scalp, face, and trunk. The father reported a history of blisters on his body when he was a child, but he was never evaluated for the problem.
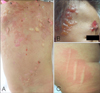
Physical examination revealed multiple, tense vesicles, bullae, erosions, and hemorrhagic crusted lesions over the face, scalp, and trunk (Fig. 1A, B). The Darier's sign was present on the thigh (Fig. 1C). There was no evidence of organomegaly or lymphadenopathy. A complete blood cell count and the biochemical profiles were within normal limits. The histological examination of a biopsy taken from the back revealed a sub-epidermal bulla with a dense infiltration of mast cells and some eosinophils in the upper dermis (Fig. 2). The toluidine blue and Giemsa stains showed that almost all of the infiltrating cells in the dermis were mast cells (Fig. 3). The direct immunofluorscence was negative. The diagnosis of DCM was made based on these clinical and histopathological findings. The patient was treated with oral levocetrizine HCl, ketotifen fumarate, and topical 0.25% prednicarbate ointment. Improvement of the symptoms was noted during a follow-up examination. Although we recommended evaluation for systemic organ involvement, the patient was transferred to another hospital based on the request of the family.
DISCUSSION
Cutaneous mastocytosis (CM) is characterized by the accumulation of mast cells in the skin without any evidence of extra-cutaneous organ involvement. CM is associated with both local and systemic symptoms that are caused by the excessive production of mast cell-dependent mediators such as histamine, leukotrienes, proteases, and/or heparin. The symptoms often vary and can include cutaneous flushing, blistering, pruritus, dyspnea, syncope, bone pain, and gastrointestinal upset including epigastric pain, vomiting, and diarrhea3,4.
CM is divided into three major subtypes: urticaria pigmentosa (UP), mastocytoma, and diffuse cutaneous mastocytosis (DCM). Approximately 58~90% of patients with CM have the UP subtype, while 10~40% of affected patients have mastocytoma5-7. DCM is the rarest subtype, accounting for only 1.74% of all cases of CM5. UP presents as yellow-tan to reddish-brown macules or slightly raised papules that are scattered over the trunk and extremities. A solitary mastocytoma presents as a brown nodule and subsequent skin lesions rarely develop. DCM may present as a diffuse reddish-brown discoloration and have a peau d'orange appearance on the entire surface of the skin. The development of systemic mastocytosis (SM) is thought to be associated with a growth factor receptor c-kit mutation and abnormal expression of cell surface adhesion antigens. However, no clear pathological mechanism has been presented that explains the development of CM8-10. Bullous eruption can be associated with all three subtypes of CM. In patients with UP, the bullous eruption usually develop during infancy and may be the presenting symptom11,12. In patients with a mastocytoma, the bulla can develop on the skin overlying the mastocytoma13. In patients with DCM, bullous eruptions are very common during the early stages of life, as seen in our patient. The blisters present in a variety of sizes and initially contain clear fluid that may become hemorrhagic with time. Bullous lesions may occur in linear or grouped fashion and often develop on the trunk, scalp, and extremities14. These lesions typically resolve by 3 to 5 years of age without scarring. The blisters seen in a patient with mastocytosis are believed to be caused by serine proteases that are released from mast cells3.
CM typically presents as a self-limiting disease, particularly in children. In about 50% of pediatric patients, the symptoms spontaneously resolve by adolescence6. Generally, DCM is seen initially almost exclusively in infants, although it may persist into adult life and has been associated with indolent systemic mastocytosis15. DCM associated with generalized bullae has a relatively poor prognosis, as this presentation has a higher rate of transformation to SM, which could cause hepatomegaly, splenomegaly, lymphadenopathy, large bone osteolysis, and anemia or pancytopenia due to bone marrow involvement6,12-14,16. DiBacco and DeLeo17 reported on eight infants that manifested bullae as their initial symptom of DCM. All of these infants exhibited systemic involvement, and two died of their disease. DCM with generalized bullae should be differentiated from other bullous diseases of childhood such as epidermolysis bullosa, staphylococcal scalded skin syndrome, incontinentia pigmenti, epidermolytic hyperkeratosis, acrodermatitis enteropathica, erythema multiforme, and toxic epidermal necrolysis.
The diagnosis of CM is based on the clinical features of the patient and the results of the histopathology. The symptoms that are caused by the release of mast cell mediators and the typical cutaneous lesions are clinically suggestive of CM. Darier's sign, the development of a wheal and erythema occurring after the brisk stroking of a lesion is also a typical finding. Abnormal proliferation of dermal mast cells on the skin biopsy specimen confirms the diagnosis. The appropriate stains used to detect mast cells in tissues include Giemsa, toluidine blue, Leder, and monoclonal antibodies that recognize tryptase or CD1174. The goal of treatment is alleviation of the symptoms and prevention of the use of potential mast cell degranulating agents or stimuli such as ingested alcohol, anticholinergic preparations, aspirin and other NSAIDs, narcotics, polymyxin B sulfate, systemic anesthetics, heat, and friction. Anti-mediator drugs such as antihistamines, cromolyn sodium, acetyl salicylic acid, and ketotifen are used in step-wise fashion to alleviate symptoms1. Cutaneous lesions may show good response to the application of psoralen plus UVA, local corticosteroids with occlusion, or intralesional injections18. Patient with extensive bullae may be at increased risk for infection. Therefore, appropriate management for preventing cutaneous infections is needed. Patients with CM, especially childhood-onset, generally have a favorable prognosis. Annual check-ups are usually sufficient for long-term management19. However, DCM with generalized bullous eruption, as seen with the patient reported here, may have a higher risk for systemic involvement and severe symptoms such as sudden death. Therefore, proper follow-up with evaluations for systemic involvement is required for these patients.


 XML Download
XML Download