

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Multiple endocrine neoplasia type 2 (MEN 2) is a rare hereditary disease and can be classified into three distinct subtypes: MEN 2A, MEN 2B and amilial medullary thyroid cancer (FMTC)1,2. Among these, MEN 2B is associated with medullary thyroid cancer (MTC), pheochromocytoma, mucosal neuroma, ganglioneuromatosis and a Marfanoid appearance3.
Mucosal neuroma is a typical phenotype of MEN 2B, one that develops mostly at birth or at around one to two years. Therefore, early detection of mucosal neuroma is a crucial part of a good prognosis and can be regarded as the dermatologic clue of diagnosis. In the domestic dermatologic literature, we found one case of MEN 2B, which was initially diagnosed as MTC at the department of internal medicine who consulted with the department of dermatology regarding the multiple papules on the lips and tongue4.
Herein, we report an interesting case of 9-year-old male who was diagnosed early as having MEN 2B by multiple mucosal neuroma and by a genetic test.
CASE REPORT
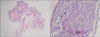
A 9-year-old male presented with asymptomatic skin-colored, verrucous surfaced papules and nodules on his lips and tongue. Since birth, the patient had lumpy papules on his lips, tongue and gingiva which gradually increased in size and number (Fig. 1). Physical examinations revealed thickened lips, an elongated face, lower jaw protrusion, large hands and feet and relatively long extremities. His length and weight were 133 cm and 27.2 kg which corresponded to growth percentiles of 60 and 47, respectively. His past medical history showed severe constipation requiring stool softener pills but the family history showed no specific endocrine disease. The complete blood cell counts, blood chemistry and thyroid function test were within normal limits. A 24-hour urine showed a vanillylmandelic acid level of 2.96 mg/day (reference range: <7.9) and a metanephrine level of 0.307 mg/day (reference range: <1.29) which also were within the normal range. However, the value of serum calcitonin was high (42.6 pg/ml) reference range: <9.9). Large amounts of fecal material in the whole colon were detected on the CT scan while the thyroid showed normal findings on a thyroid ultrasonogram. Histopathological examination of tissue biopsies from his lips and tongue showed multiple dermal nodules caused by hypertrophy of mucosal nerves. Schwann cells formed broad, uniform, interlacing fascicles in asymmetric patterns and there were scattered nuclear palisades. But atypical mitotic figures and nuclear pleomorphism were not found (Fig. 2). The immunohistochemical profile of nerve fascicles was positive for S-100 protein and surrounding capsules were immunoreactive for epithelial membrane antigen (EMA) (Fig. 3). In DNA analysis, sequencing of exon 16 in the RET proto-oncogene revealed a missense mutation, where ATG was substituted by ACG at codon 918 (Met918Thr) (Fig. 4). Based on his clinical, histologic and genetic features, we diagnosed the patient as MEN 2B and recommended total thyroidectomy. However, further follow up was not done.
DISCUSSION
MEN 2 is an autosomal dominant hereditary disease that is classified into three distinct subtypes1,2. Though there are some variations among reports, MEN 2A accounts for about 75% of all MEN 2 cases and expresses MTC, pheochromocytoma and parathyroid gland hyperplasia3,5. FMTC is another variant which accounts for about 20% of MEN 2 cases and has a particularly benign course of MTC and a low incidence of other clinical manifestations5. MEN 2B occupies only 5% of MEN 2 cases. However, its clinical course is the most aggressive one5. Though MEN 2B is similar to MEN 2A, mucosal neuroma, ganglioneuromatosis of the intestinal tract and Marfanoid habitus can be seen only in MEN 2B with parathyroid gland hyperplasia being rare5-7.
Mucosal neuroma is the most characteristic clinical phenotype and the earliest sign of MEN 2B and develops at birth or at around one to two years in almost all MEN patients4. Mucosal neuroma generally develops in the lips, tongue and buccal mucosa and less commonly in the palate, intestinal mucous membrane and conjunctiva8. As time goes by, mucosal neuromas can increase in size and number or show no change. Because it has no specific symptoms and no malignant changes, no further treatment is needed except for cosmetic purposes. Our patient also had multiple papules and nodules on his lips and tongue when he was born and the size and the number increased gradually as he got older without any irritation history. Chronic constipation caused by the intestinal ganglioneuromatosis and Marfanoid habitus are also early signs of MEN 2B like mucosal neuroma9-11. Our patient also had suffered from severe constipation. Therefore, he had taken stool softener pills intermittently since infancy. Additionally, our patient showed a Marfanoid habitus such as lower jaw protrusion, above average height, long slender limbs and flat feet.
MTC commonly develops in all subtypes of MEN 2 and is the most important prognostic factor. Usually, MTC develops relatively young, exhibits a more aggressive disease course, and accounts for more than 95% of MEN 2B cases2,3,5. In particular, MTC is resistant to chemotherapy or radiotherapy if it spreads to another site by metastasis. Therefore, early diagnosis and prophylactic total thyroidectomy can minimize the disease course and mortality rate. Though our patient exhibited normal results on a thyroid function test and an ultrasonogram, his serum calcitonin increased by 42.6 pg/ml and a mutation M918T was confirmed by genetic testing. To ensure a favorable outcome, a prophylactic thyroidectomy was done.
MEN 2B is caused by germline missense mutations in the RET proto-oncogene. The RET gene which is located on chromosome 10q11.2 encodes a receptor tyrosine kinase. It is expressed in neuroendocrine cells including thyroid C cells, urogenital system cells, adrenal glands, and parasympathetic and sympathetic ganglia. It plays an important role in cell growth and differentiation3,5. More than 90% of MEN 2B cases are caused by a single point mutation of M918T at 918 codon in exon 16 of the RET gene. The others are caused by an A883F substitution in the 883 codon in exon 15 or a compound heterozygous mutation of V804M with Y806C or V804M with S904C12-16. Unlike MEN 2A, most MEN 2B cases are caused by de novo mutations of RET gene. Therefore, most patients do not have a family history3. In our case, a missense mutation of ATG to ACG was identified. This was thought to have developed sporadically with the patient not having a family history. Most of all, genetic counseling for other family members are needed.
In conclusion, MEN 2B is often caused by de novo mutations of the RET proto oncogene. Therefore, DNA analysis is necessary for confirmation. However, it is hard to do routine DNA analysis in newborns, except for patients who have a family history of MEN 2B. Therefore, mucosal neuroma, a distinctive manifestation of MEN 2B (as in our patient), can be the definitive early diagnostic clue. Hence, the dermatologist should play a key role in pursuing further genetic investigations and prompt treatment.



 XML Download
XML Download