

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Lymphomatoid papulosis (LyP) is a rare disorder characterized by recurrent papulonodular cutaneous lesions1. The clinical course varies: 10% to 20% of adult patients with LyP have associated lymphomas such as mycosis fungoides, T-cell immunoblastic lymphoma, or Hodgkin's disease, which can precede, appear simultaneously with, or develop after the diagnosis of LyP2. Though well documented in adults, LyP is uncommon in children3, and few cases have been reported in pediatric patients who developed lymphoma4,5. We report the case of a 12-year-old boy, who developed anaplastic large cell lymphoma (ALCL) on the scrotum, 13 months after identification of papulonecrotic lesions of LyP on both lower extremities and face. This is the first report of such as case in the Korean medical literature.
CASE REPORT
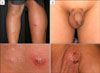
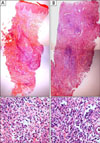
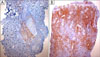
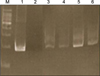
In August 2007, an 11-year-old boy was referred to our department for the evaluation of multiple, erythematous papules located predominantly on the face and both legs (Fig. 1A). He was otherwise in good health. A skin biopsy from the left knee showed a folliculocentric distribution that an admixture of various cell types, including scattered atypical large lymphoid cells with large vesicular nuclei, eosinophils and small lymphocytes, which are typical of type A LyP (Fig. 2A). After several months, these lesions resolved spontaneously without any treatment. In September 2008, the patient was referred again to our department after a similar skin lesion was noted five days previously. Skin examination revealed a tender, solitary, 1.5 cm sized, subcutaneous nodule with central erosion and a hemorrhagic crust on the left scrotum (Fig. 1B). Histological examination showed that the deep dermis and the subcutaneous fat tissue were extensively involved with more prominent proliferation of large atypical cells, including many atypical mitoses (Fig. 2B). The immunohistochemical staining of the atypical cells on both specimens were essentially identical and were as follows: leukocyte common antigen (+), ubiquitin carboxyl-terminal esterase 1 (+), CD4 (+), CD8 (-), CD30 (+), and anaplastic lymphoma kinase (ALK) (-) (Fig. 3). The rearranged bands of the T cell receptor (TCR) gene rearrangement were identical in both specimens (Fig. 4). The physical examination, thoracic and abdominal computed tomography (CT) showed no evidence of lymphadenopathy; however, focal bone involvement of the lymphoma at the right ischium was detected on magnetic resonance imaging and positron emission tomography/CT. But bone-marrow biopsy revealed no abnormalities. We diagnosed cutaneous ALCL involving bone. Since then, the patient has been treated with four cycles of intensive chemotherapy using the modified NHL-BFM-90 protocol (AA-BB-CC-AA cycles; AA: MTX, ifosfamide, VP-16, and cytarabine, BB: vincristine, cyclophosphamide, MTX and doxorubicine, CC: cytarabine and vindesine). To date, mucositis has occurred once without other serious complications, and the lymphoma has regressed in the skin and bone lesions.
DISCUSSION
According to the 2008 World Health Organization (WHO) classification6, LyP and ALCL are categorized as mature T-cell neoplasms, and two major groups of ALCL have been described. One group is LyP and primary cutaneous ALCL, which is listed as a single nosologic entity within the spectrum of CD30+ lymphoproliferative disorders, and the other group is systemic nodal ALCL, which is divided into ALK+ and ALK- types. In general, systemic ALCL is ALK+, and is usually more aggressive and characterized by infiltration of other organs. By contrast, primary cutaneous ALCL is ALK+ and rarely involves extracutaneous organs. Therefore, if a lymphoma with ALK-, CD30+ anaplastic large cells initially develops in the skin with subsequent bone involvement, primary cutaneous ALCL and systemic ALK- ALCL need to be differentiated. However, it is difficult to distinguish primary cutaneous and systemic ALCL because reliable criteria are currently unavailable. Because of the absence of distinctive features of these diseases by histological and immunohistochemical analysis further clinical examination is needed to confirm the diagnosis7,8. Despite the bone involvement in our case, systemic ALCL involving skin did not seem the correct diagnosis because the thoracic and abdominal CT showed no evidence of lymph node involvement. Moreover, systemic ALK- ALCL usually occurs in older individuals and has a poor prognosis. Therefore, the diagnosis of primary cutaneous ALCL with bone involvement was established. The absolute confirmation of our diagnosis can be made only after careful monitoring of our patient over many years.
Despite its chronic course, LyP is generally known to have a favorable prognosis. In some cases, however, the development of other lymphomas has been reported. Furthermore, Brown and Skarin9 reported that 20% to 80% of patients with LyP progress to lymphoma with others reporting that lymphoma can occur prior to, simultaneously with, or after the manifestation of LyP4,5,10,11. However, cases of pediatric LyP patients developing a malignancy are extremely rare (Table 1)4,12-14. Of particular note, there has only been one case of lymphoma developing after LyP diagnosis in a pediatric patient11. The patient in question was diagnosed with diffuse large-cell lymphoma three years after the onset of LyP. The patient was treated with radiation for tumor regression and had no recurrence for ten years. In our case, the progression of LyP to lymphoma had occurred in a relatively short period of time and the tumor regressed after chemotherapy.
Genotype analysis of LyP and lymphoma have been performed to demonstrate their association. Weiss et al.15 analyzed the DNA from six patients diagnosed with LyP for rearrangement of the beta and gamma TCR genes. Five patients had a clonal rearrangement, and three patients had lymphoma involvement. This finding therefore suggests that malignant transformation might have occurred in the LyP. In addition, the clonal expansion of the TCR gene in the LyP lesions indicates an association between the LyP and the ALCL.
Immunophenotypic and genotypic analysis of LyP and ALCL lesions from patients with both diagnoses have revealed identical T-cell clones4. These results indicate that an additional malignant transformation of LyP developed or a common malignant T-cell clone proliferated over the course of both the LyP and ALCL lesions. However, it is still difficult to explain why the LyPs and lymphomas with the same T-cell clone have different clinical course. Amagai et al.5 reported that LyP and ALCL have different characteristics, despite their identical immunophenotypes and genotypes. This biological difference might be caused by additional mutations in growth-related genes in LyP cells that become transformed into lymphoma cells. In addition, it is possible that immunosurveillance changes the biological behavior of the lymphocytes in the skin. We have not confirmed whether identical clones proliferated in each of the lesions associated with ALCL and LyP in our patient. However, the histological changes and TCR gene rearrangement in our case support the previously proposed hypothesis that ALCL and LyP represent steps along a continuous spectrum. In addition, our case illustrates the different clinical manifestations of proliferation of the same clones with regard to ALCL and LyP.
Despite recent studies that have identified risk factors that cause progression of LyP to lymphoma, current knowledge remains incomplete. According to retrospective research16, men with LyP develop lymphoma 2.5 times more frequently than women, although no other significant differences risk factors have been identified. Also, the median and maximum times to develop lymphoma have been reported to be 3.04 years and 40.89 years, respectively. Close and regular follow-up of patients is therefore important.
The prognosis for primary cutaneous ALCL in children is unknown. This is because primary cutaneous ALCL is a rare tumor in children and infiltration into the bone is extremely rare17. Ng et al.18 reported that the overall outcome of primary bone ALCL is poor, with over 60% of cases dying within two years of diagnosis despite intensive therapy, compared to a favorable outcome in cases with nodal and non-bony extra nodal disease. By contrast, Williams et al.19 reported that patients with bone lesions did not have a worse prognosis. The 5 year event-free survival in 9 patients with bone involvement was 78%. However it is unclear whether they presented with primary bone ALCL or ALCL with metastatic bone disease. In our case, intensive chemotherapy was relatively effective, and resulted in a good response. However, it was difficult to assess the curative and prognostic effects since the follow-up period was limited. Therefore, long term follow up is required.
To the best our knowledge, this is the first reported case of LyP followed by ALCL in the Korean literature and the first reported case of LyP followed by lymphoma involving both skin and bone in the English literature. While LyP is a benign disease that does not require treatment, it can progress to lymphoma. Therefore, we strongly emphasize the importance of close follow-up of patients with LyP to monitor for progression to lymphoma.

 XML Download
XML Download