

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Erdheim-Chester disease (ECD) is a rare disease first described in 19301. There are no definitive diagnostic criteria for this entity, and diagnosis is usually based on radiologic findings of osteosclerosis combined with histopathological evidence of foamy histiocytic infiltration1. ECD can affect multiple organs and shows various clinical manifestations, depending on the organs involved, ranging from asymptomatic disease to respiratory distress and/or cardiac failure2.
We describe here a patient with ECD with multi-organ involvement, including the skin, bones, pituitary gland, pericardium, and retroperitoneum.
CASE REPORT
A 68-year-old man was referred for evaluation of skin lesions on his face and scalp. He had been hospitalized in the endocrinology department for central diabetes insipidus and was being treated with desmopressin therapy. He had recently complained of coughing and dyspnea.
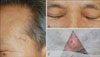
Skin examination revealed multiple, irregularly-shaped yellowish plaques on the left temple, forehead, and both lower eyelids, which had progressed for 1.5 years (Fig. 1A, B). Apart from his facial lesions, we found a well-demarcated, 1 cm-sized, pinkish nodule on the right parietal scalp, which had been present for 4 years (Fig. 1C). All skin lesions were asymptomatic. Histopathological examination of skin lesions from the left temple and scalp revealed dermal infiltration of foamy histiocytes, along with variable numbers of multinucleated giant cells and lymphocytes, favoring diagnoses of plane xanthoma and xanthogranuloma, respectively (Fig. 2).
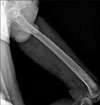
Laboratory tests showed a slightly increased erythrocyte sedimentation rate and leukocytosis, suggesting an inflammatory condition, but renal and liver function tests and lipid profiles were normal. We suspected ECD, and therefore evaluated skeletal involvement despite the absence of a history of bone pain. Plain radiography of the left femur showed diffuse osteosclerotic changes with focal osteolysis in the diaphyseal and metaphyseal regions of the left distal femur (Fig. 3). Technetium-99m bone scintigraphy showed increased symmetric uptake by many bones, including the skull, ulnae, femurs, tibiae, calcanei, ribs, and hip bones (Fig. 4), which suggested bilateral symmetric changes in multiple bones. 18F-fluorodeoxyglucose positron emission tomography confirmed the presence of symmetric hypermetabolic lesions in many bones, as well as multifocal hypermetabolic infiltrative lesions in the perirenal space and around the aorta, accompanied by pericardial and pleural effusion. Immunohistochemical examination of perirenal tissue obtained by laparoscopic biopsy showed that the infiltrating histiocytes were CD68-positive and S100-negative, and showed evidence of fibrosis as well (Fig. 5). Based on the radiological and histopathological findings, the patient was diagnosed with ECD with involvement of the skin, bones, pituitary gland, pericardium, and retroperitoneum. Treatment with interferon-alpha (6 million units subcutaneously three times per week) was initiated, and the treatment has been well tolerated for 16 months. Echocardiography showed a decreased amount of pericardial effusion and the patient's clinical condition has remained stable.
DISCUSSION
ECD is a rare non-Langerhans cell histiocytosis, characterized by infiltrates of foamy histiocytes in conjunction with symmetric osteosclerotic changes in the long bones. The etiology and pathogenesis of ECD remain unclear, although abnormal activation of monocytes may be involved3.
The clinical manifestations of ECD vary tremendously, as the symptoms depend on the tissues infiltrated. A review of 59 ECD patients found that bone pain, predominantly involving the lower limbs, was the most frequent symptom (47% of patients), and that extraskeletal manifestations, including exophthalmos, diabetes insipidus, and retroperitoneal histiocytic infiltration, were not uncommon (30% of patients)1. In addition, 19% of ECD patients showed skin involvement, predominantly of the eyelids1 as in our case. Histopathological examination is crucial for accurate diagnosis, especially to differentiate ECD from other types of histiocytosis. The majority of infiltrating cells in ECD are foamy histiocytes, frequently associated with variable amounts of fibrosis and the presence of inflammatory cells such as lymphocytes, plasma cells, and Touton-type giant cells4. Immunohistochemically, most histiocytes in ECD are negative for S-100 and positive for CD68, indicating that these cells are from the macrophage, and not the dendritic cell lineage.
Apart from histological findings, the only specific signs of ECD are radiological findings in the long bones, such as symmetrical sclerosis of the metaphyses and diaphyses of the long tubular bones1. These features differentiate ECD from Langerhans cell histiocytosis (LCH), in which the bone lesions are usually osteolytic and rarely involve the long bones5. In some patients with ECD, clinical bone symptoms can be mild or absent. Thus, bone scintigraphy may be useful for detecting bone lesions, as in our case. Although our patient did not complain of bone pain, the most common symptom of ECD, his history of diabetes insipidus and skin lesions suggested a diagnosis of ECD which was confirmed by the results of bone scintigraphy and retroperitoneal biopsy.
An optimal treatment for ECD has yet to be established, probably because of the rarity of the condition and the paucity of clinical trials. Systemic steroids, various cytotoxic agents, radiation therapy, and hematopoietic stem cell transplantation have all been used to treat patients with this condition, with variable outcomes6,7. At present, interferon alpha is used as first-line treatment of ECD8. Indeed, we have now treated our patient with interferon alpha for 16 months without serious complications. Although the clinical symptoms of our patient seem to be stable, the efficacy of interferon alpha has been reported to be inconsistent or limited, especially for cardiovascular, cerebral, and mesenteric lesions8,9. Therefore, close long-term follow-up will be required to evaluate the efficacy of interferon alpha in our patient, and efforts should be made to establish novel effective therapeutic regimens.
The prognosis of patients with ECD has been reported to be dismal, with a mean overall survival period of 32 months and a higher mortality rate than that for patients with LCH (57% versus 30%)1. The most commonly reported causes of death in patients with ECD include respiratory and heart failure, and involvement of the bones and soft tissues has been associated with poor prognosis.
Although ECD is a rare disease, approximately 350 such patients have been described to date. The true incidence of the disease may be much higher and lack of knowledge of the condition and difficulties in diagnosis may contribute to the apparent low incidence. Increased awareness of the condition may enhance prompt diagnosis and appropriate management.



 XML Download
XML Download