

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Kasabach-Merritt syndrome (KMS) is a rare complication of pre-existing hemangioma in infancy and early childhood1. Patients with KMS present with an enlarged hemangioma and laboratory features of thrombocytopenia and consumptive coagulopathy. KMS is associated with several types of hemangiomas in infancy, including kaposiform hemangioendothelioma (KHE), tufted angioma, and infantile hemangioma. Among those hemangiomas, KHE occurs most frequently before KMS2. Tufted angioma is a benign vascular tumor characterized by histopathological features of tufts of capillary-sized vessels within the dermis3. Although a recent study showed spontaneous regression in most cases within 2 years4, these benign tumors may develop potentially fatal KMS as complication2,3.
With an overall mortality rate of 12%, various treatment regimens have been introduced to manage KMS5. Recent reports recommended a careful stepwise approach in the management of KMS, but the results demonstrated inconsistent efficacy6,7. Among the regimens recommended, systemic corticosteroid is considered as first-line therapy for KMS patients because of its low cost and ease of administration6-8. We report a case of KMS arising from tufted angioma successfully treated with systemic corticosteroid.
CASE REPORT
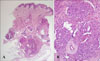
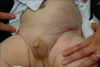
A 2-month-old male infant presented with a skin lesion on his left pubis 2 weeks after birth. He had no complications during birth. Physical examination revealed a solitary, 2.5×2.5 cm-sized, erythematous induration on his left pubis (Fig. 1A). A 3 mm punch biopsy was performed at the center of the lesion. Histopathological examination demonstrated well-circumscribed lobules composed of capillaries resembling "cannonballs" within the dermis. The lobules were composed of small dilated capillaries surrounded by crescent-shaped vascular structures. The lesion was diagnosed as tufted angioma (Fig. 2).
One month following skin biopsy, the lesion suddenly expanded to the abdomen and scrotum (Fig. 1B). Physical examination revealed a purpuric and firm induration covering the lower half of the left abdomen, pubis, scrotum and upper to medial thigh. The initial laboratory work-up revealed severe thrombocytopenia (platelet counts, 61,000/mm3); increased D-dimer (15,335 ng/ml, reference range: 0~243 ng/ml); increased fibrin degradation products (1:8~16 positive, reference: 1:2 negative) and decreased fibrinogen (67 mg/dl, reference range: 200~400 mg/dl), suggestive of consumptive coagulopathy. Abdominal ultrasound imaging study revealed mild hepatomegaly. Other laboratory results including white blood cell counts, hemoglobin, bleeding time, coagulation time, prothrombin time (PT), antithrombin III, and activated partial thromboplastin time (aPTT) were within the normal range. He was diagnosed with KMS arising from a pre-existing tufted angioma based on the clinical findings and laboratory values. The patient was admitted to commence immediate treatment with intravenous dexamethasone at 0.32 mg/kg/day (equivalent to prednisolone 2.0 mg/kg/day). Two days after initiating therapy, his platelet counts recovered to 222,000/mm3 and the lesion ceased to expand. Subsequently, intravenous dexamethasone was changed to oral prednisolone, then gradually tapered to 1.5, 1.2, 1.0 mg/kg/day over 15 days and finally to 0.5 mg/kg/day over two weeks. The wide purpuric induration was shrunken to a small bluish, erythematous induration (Fig. 3). Then, treatment was stopped and the patient showed no signs of recurrence for a year. There were no remarkable complications related to systemic corticosteroid during the treatment and follow up period.
DISCUSSION
KMS is a rare complication of pre-existing hemangiomas first described by Kasabach and Merritt1 in 1940. It is characterized by an enlarging hemangioma, associated with thrombocytopenia and consumptive coagulopathy which may lead to life-threatening bleeding1. More than 80% of cases occur within the first year of life9. KMS can be a complication of KHE, tufted angiomas or, rarely, large infantile hemangiomas but most reported cases of KMS were associated with KHE and tufted angiomas2. A recent retrospective study conducted by Enjolras et al.10 revealed that KMS was due either to KHE and/or tufted angiomas. The residual lesions of patients with cured KMS were predominantly tufted angiomas, whereas KHE was more common during the active phase of KMS. Thus it is possible that KHE and tufted angiomas are the part of the same neoplastic spectrum and histologic continuum11. Enjolras et al.10 proposed that KMS was not a complication of infantile hemangiomas, but of KHE and tufted angiomas based on their findings.
The trigger factors for the development of KMS include surgical intervention, pregnancy, angiography, and needle aspiration of hemangioma12-14. In our case, KMS occurred one month after 3 mm skin biopsy from a pre-existing tufted angioma. Minor trauma due to punch biopsy might have triggered KMS in our case.
The pathogenesis of KMS remains unestablished. However, platelet trapping by abnormally proliferating endothelium within the hemangioma has been proposed as a possible mechanism15. Platelet trapping can result in the activation of platelets with secondary activation of coagulation cascades, eventually leading to consumption of various clotting factors. Immunohistochemical study using monoclonal antibody against CD61, a marker of platelets, and isotope studies using 111indium-labeled platelets and 51Cr-labeled platelets support the possible role of platelet trapping in the development of KMS15. The reason why platelets are trapped within the hemangioma in the patients with KMS remains unelucidated. In addition to platelet trapping, excessive blood flow and sheer stress secondary to arteriovenous shunts within the tumors may cause further platelet activation. Both thrombocytopenic status and reduction in coagulation factors eventually result in bleeding within the tumors that manifests as a rapidly growing hemangioma.
Management of KMS has been challenging because of its rarity and there are no well-established systematic treatment strategies to date. Systemic corticosteroid is considered to be first-line therapy in patients with KMS (Table 1)6-8,15-18. One study demonstrated that treatment with prednisolone at 2 or 3 mg/kg/day achieved response in 30% to 72% of infants16. However, other data suggested variable response to prednisolone, ranging from no efficacy to significant regression in lesion size, and even complete remission7. The discrepant results may be due to variation in patient clinical characteristics in the studies, such as different pre-existing tumors, and the possibility that the enrolled patients actually had vascular malformations, rather than hemangiomas. If a lesion responds to steroid, the dose should be reduced slowly to prevent recurrence. High doses of prednisolone (more than 5 mg/kg/day) can be used as an alternative, and it has shown to be more effective than 3 mg/kg/day in a study by Sadan and Wolach17. Some authors advocate the superiority of high dose oral methylprednisolone therapy to the conventional treatment with prednisolone18. High rates of initial response and short therapy duration appears to be an advantage, but should be confirmed by a double blind, comparative study between different regimens. The mechanisms of prednisolone in controlling thrombocytopenia, coagulopathy, and eventually stabilization of hemangiomas remain unclear, although it appears to increase platelet longevity, increase vasoconstriction, inhibit fibrinolysis, and disrupt angiogenesis. Side-effects associated with steroid treatment include hypertension, cushingoid appearance, growth suppression and opportunistic infections15,19. Our patient commenced with 0.32 mg/kg/day of intravenous dexamethasone, equivalent to 2.0 mg/kg/day of prednisolone. The most likely cause of our patient's rapid platelet recovery and lesion stabilization following initiating therapy was the superior bioavailability of intravenous steroids over that of oral administration.
In cases resistant to systemic corticosteroid, multiple treatment modalities can be used in a stepwise manner, including interferon-alpha, an antiproliferative and antiangiogenic agent, and vincristine, a strong inhibitor of angiogenesis. Radiotherapy is another option which can induce embolization within the hemangiomas. These options are usually recommended as second-line therapies6,7,15. However, treatment with these regimens can result in severe adverse events. The known adverse effects of interferon therapy include flu-like symptoms, neutropenia, and spastic diplegia in infancy5,7. Vincristine can be associated with transient peripheral neuropathy, and it is considered a relatively safe regimen with high efficacy7,8,15. Since radiotherapy has been reported to induce late side effects, including local growth failure, late skin changes, destruction of bone tissue and secondary malignancies, it should be administered to patients with life-threatening lesions, or to patients with lesions that may compromise organ function, or when other therapies fail to achieve response5-7,15.
In conclusion, conventional systemic corticosteroid is probably the most cost-effective treatment option in patients with KMS. Systemic corticosteroid may result in variable response in KMS; however, it should be considered first-line treatment since it offers rapid resolution and less complications, compared to other treatment modalities. In addition, intravenous corticosteroid may be more efficacious than oral corticosteroid, most likely due to superior bioavailability. However, due to the rarity of this syndrome, it is difficult to undertake any comparative studies on different modalities.


 XML Download
XML Download