

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cholesterol is a major lipid component of human skin. The biosynthesis of cholesterol is in tight association with the cutaneous barrier function1. Cholesterol, a product of the mevalonate isoprenoid pathway, is also essential for cellular functions, such as cell growth, cytokinesis, and differentiation2,3. Indeed, cholesterol plays a number of critical roles in cellular function. Having the appropriate amount of cholesterol in the appropriate place is essential for membrane structure, signal transduction, and overall human health. At the cellular level, cholesterol increases membrane thickness and decreases transbilayer permeability4,5.
Cholesterol regulates both the flexibility and the mechanical stability of the membrane bilayer and is involved in receptor-mediated signaling6-9. Cholesterol plays a critical role in assembling membrane microdomains, such as lipid rafts and caveolae9. Depletion of the plasma membrane cholesterol causes increased binding of EGF to EGF receptor, increased dimerization of the EGFR, and hyperphosphorylation of the EGFR. Addition of cholesterol reduces EGF binding to EGFR and EGF-induced EGFR activation10. Thus, cholesterol appears to have regulatory effects on receptor tyrosine kinase-mediated signaling9-11. In addition, cholesterol depletion by βCD and methylbeta-cyclodextrin (MβCD) induces apoptosis of HaCaT cells through activation of caspase-8, and βCD-induced apoptosis is accompanied by mitochondrial cytochrome c release12.
Matrix metalloproteinases (MMPs) cleave collagens and other components of the extracellular matrix and play an important role in physiological processes of tissue remodeling. Matrix metalloproteinases can be divided into the following subgroups: collagenases, gelatinases, stromelysins and stromelysin-like MMPs, matrilysins, membrane-type (MT) MMPs and other MMPs13,14.
MMP-1 is a key enzyme for degradation of extracellular matrix such as procollagen type I. Expression of the interstitial collagenase (MMP-1) is mediated through activator protein-1 (AP-1)15,16. The proximal AP-1 element located between -72 and -66 of MMP-1 promoter region plays a major role in the transcriptional regulation of MMP-1 gene expression16-18. The MAP kinase family, which includes ERK, JNK and p38, is known to have an important role in signaling processes which regulate MMP-1 gene expression19,20. UV irradiation has been reported to activate ERK and JNK/p38 kinase, and these activated MAP kinases increase the expression of c-Fos and c-Jun, which are the main components of the AP-1 transcription factor21,22. Binding of activated AP-1 to its response element on the MMP-1 promoter will increase the transcription of MMP-123.
In this study, we investigated whether cholesterol regulates MMP-1 expression and, if so, how does cholesterol regulate MMP-1 expression in cultured human dermal fibroblasts. Our results demonstrated that the amount of intracellular cholesterol regulates ERK and JNK phosphorylation, which may play a pivotal role in the induction of MMP-1 expression in human dermal fibroblasts.
MATERIALS AND METHODS
Reagents
Cell culture media (Dulbecco's modified Eagle's medium, DMEM), antibiotics, and TRIzol reagent were purchased from Life Technologies (Rockville, MA, USA). Fetal bovine serum (FBS) was obtained from Hyclone (Logan, UT, USA). Rabbit polyclonal anti-p-ERK1/2, anti-p-SAPK/JNK, anti-p-p38, anti-ERK1/2, anti-SAPK/JNK, and anti-p38 were purchased from Cell Signaling Technology (Beverly, MA, USA). Mouse monoclonal anti-MMP-1 was from Oncogene (San Diego, CA, USA). The cholesterol assay kit was from ShinYang (Seoul, Korea). MβCD, cholesterol and filipin were from Sigma (St. Louis, MO, USA). MEK inhibitor, U0126, JNK inhibitor, SP600125 and p38 inhibitor, SB203580 were purchased from Calbiochem (San Diego, CA, USA).
Cell cultures
Primary cultures of human dermal fibroblasts were obtained from healthy donors (age 20~30 y). The skin was minced, followed by incubation with collagenase (1 mg/ml in DMEM) for 1~2 h at 37℃. Collagenase was then removed by washing with DMEM. The isolated cells were allowed to attach on plastic plates and cultured in DMEM supplemented with 10% FBS, 2 mM glutamine, 100 IU/ml penicillin, and 100µg/ml streptomycin. After six to eight passages, the fibroblasts were used for experiments.
MβCD and cholesterol treatments
For experiments, fibroblasts were maintained in culture medium without FBS for 24 h; and then, 10 mM MβCD and/or 100µg/ml cholesterol was added for 1 h. Culture media were replaced with fresh media without FBS and the cells were further incubated for the indicated times. In experiments involving MAPK inhibitors, these were added 30 min prior to 10 mM MβCD and/or 100µg/ml cholesterol. Normal human dermal fibroblasts were used in three independent experiments.
Cholesterol assays
Cellular cholesterol assay was performed using a commercial kit according to the manufacturer's instructions. Briefly, cells were washed twice with 1 ml of cold PBS, and lipids were extracted with 2 ml of extraction buffer [hexane/isopropyl alcohol (3:2, v/v)] for 1 h at room temperature. The organic extract was removed from the cell monolayer, transferred to glass tubes and the solvent was removed in a SpeedVac. The lipid was solubilized in 1 ml of the cholesterol assay kit buffer solution and vortexed to solubilize the lipid pellet. Samples were incubated for 1 h at 37℃ prior to measuring absorbance at 505 nm.
Filipin staining
Filipin is fluorescent and is widely used to localize cellular cholesterol24. Cells were grown on polylysine-coated coverslips, rinsed with cold PBS, and fixed in 4% paraformaldehyde (Merck, Darmstadt, Germany) on ice for 30 min. Cells were then rinsed three times with PBS for 10 min, and were treated with 100µg/ml filipin for 2 h at room temperature. Cells were again rinsed thoroughly and then viewed by a fluorescence microscope using a UV filter set (Olympus Venox AHBT3/Q imaging system, Tokyo, Japan).
Immunoblotting
Cells were lysed with lysis buffer [150 mM NaCl, 10 mM Tris, (pH7.4), 1 mM EDTA, 1 mM EGTA (pH8.0), 1% Triton X-100, 0.5% NP-40, 0.2 mM PMSF, protease cocktail, phosphatase cocktail] and lysates were clarified by centrifugation at 13,000 rpm for 15 min at 4℃. Extracted protein was quantified by the Bradford assay. Proteins were separated on 8~16% SDS-PAGE and transferred to PVDF membranes (Amersham Bioscience, Buckinghamshire, UK). After transfer, PVDF membranes were stained with Ponceau S (Sigma, St. Louis, MO, USA) and blocked with TBS containing 5% skim milk at room temperature, then incubated overnight at 4℃ with individual antibodies against MMP-1, ERK1/2, JNK, or p38, and then incubated with horseradish peroxidase-conjugated rabbit anti-mouse antibody and donkey anti-rabbit antibody, respectively. The ECL western blotting system (Amersham Bioscience, Buckinghamshire, UK) was used for protein detection as per the suggestion.
Reverse transcription-polymerase chain reaction
Total RNA from cells was extracted by using the TRIzol reagent (Life Technologies, Inc., Rockville, MD, USA) according to the manufacturer's protocol. Isolated RNA samples were then used for reverse transcription-polymerase chain reaction (RT-PCR). Samples of 1µg total RNA were reverse transcribed into cDNA in 20µl reaction volumes using a first-strand cDNA synthesis kit for RT-PCR according to the manufacturer's instuctions (MBI Fermentas, Hanover, MD, USA). Semi-quantitative PCR was performed with 1µl of the first-strand cDNA product using the following primers from human genes: MMP-1 (forward, 5'-ATT CTA CTG ATA TCG GGG CTT TGA-3'; reverse, 5'-ATG TCC TTG GGG TAT CCG TGT AG-3'). and GAPDH (forward, 5'-ATT GTT GCC ATC AAT GAC CC-3'; reverse, 5'-AGT AGA GGC AGG GAT GAT GT-3') The PCR conditions used were: one cycle of initial denaturation (for 5 min at 94℃), 28 cycles (MMP-1) or 21 cycles (GAPDH) of amplification (for 1 min at 94℃, for 1 min at 60℃, and for 1 min at 72℃) and one cycle of final extension (for 10 min at 72℃)25. The PCR amplifications were performed in cycle numbers corresponding to the logarithmic amplification phase. The reaction products were electrophoresed in 2.0% agarose gels and visualized with ethidium bromide. The signal strengths were quantified using a densitometric program (TINA; Raytest Isotope-nme[β]gerate, Germany).
Electrophoretic mobility shift assays
Electrophoretic mobility shift assays (EMSA) were performed using a commercial kit, according to the manufacturer's instructions (Promega, Madison, WI, USA). Briefly, AP-1 (5'-CGC TTG ATG CAG CCG GAA-3') consensus oligonucleotides were end labeled by T4 polynucleotide kinase using [γ-32P] ATP (3,000 Ci/mmol; Amersham Pharmacia Biotech., Piscataway, NJ, USA). Binding reactions were performed for 30 min on ice with 5µg of protein in 20µl of binding buffer containing 4% glycerol, 20 mM HEPES (pH 7.9), 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT, 50 mM NaCl, 10 mM Tris-HCl (pH 7.5), 50µg/ml poly (dI-dC) and 20,000~25,000 dpm of 32P-labeled oligonucleotide. DNA-protein complexes were separated from unbound oligonucleotide by electrophoresis through 6% DNA retardation gels (Invitrogen, Carlsbad, CA, USA) using 0.5x Tris-borate.
RESULTS
Cholesterol treatment decreased the expression of MMP-1 mRNA and protein in cultured human dermal fibroblasts
To determine whether cholesterol directly regulates MMP-1 expression in cultured human dermal fibroblasts, the cells were treated with the indicated concentration of cholesterol for 1 h. The cells were harvested after 24 h of cholesterol treatment to observe the changes in MMP-1 mRNA expression by RT-PCR, and culture media were harvested 72 h post-treatment to measure MMP-1 protein expression by Western blot analysis. Cholesterol treatment significantly decreased the MMP-1 mRNA (Fig. 1A) and protein (Fig. 1B) expression in a dose-dependent manner. The expression of MMP-1 mRNA decreased significantly to 60.5±4.2% and 37.8±4.4% of the control level with 50µg/ml and 100µg/ml of cholesterol, respectively (Fig. 1A). The level of MMP-1 protein also decreased significantly to 39.2±8.4%, 40.1±10.2%, 16.5±5.3%, and 10.4±6.2% of the control level with 5µg/ml, 10µg/ml, 50µg/ml and 100µg/ml of cholesterol, respectively (Fig. 1B). Cell viabilities measured by MTT assays were not affected by cholesterol treatment (data not shown).
Cholesterol depletion increased the expression of MMP-1 mRNA and protein through ERK and JNK/AP-1 pathway in cultured human dermal fibroblasts
We also examined the effects of cholesterol depletion on the expression of MMP-1 mRNA and protein in cultured human dermal fibroblasts. The cholesterol depletion agent, MβCD, was added to the culture media for 1 h at the indicated concentration and then the cells were harvested at 24 h after MβCD treatment to observe changes in MMP-1 mRNA expression by RT-PCR, and the culture media were harvested 72 h post-treatment to measure the MMP-1 protein expression by Western blotting. MMP-1 mRNA expression increased significantly by an average of 10, 30, 32 and 35 folds with 2 mM, 5 mM, 10 mM and 20 mM of MβCD, respectively, in human dermal fibroblasts (Fig. 2A). MMP-1 protein expression also increased significantly by an average of 4, 4.5, 9 and 20 folds with 2 mM, 5 mM, 10 mM and 20 mM of MβCD, respectively (Fig. 2B). On the other hand, we also confirmed that MMP-1 expression was significantly increased by HMG-CoA reductase inhibitor, fluvastatin treatment in human dermal fibroblasts (data not shown).
To investigate the molecular signaling pathways involved in cholesterol depletion-induced MMP-1 expression in cultured human dermal fibroblasts, we studied the effects of cholesterol depletion and repletion on the activation of MAP kinases, including ERK1/2, JNK and p38 kinases. Cells were treated with 10 mM MβCD with or without 100µg/ml cholesterol for the indicated times. Cholesterol depletion by MβCD treatment increased phosphorylation of ERK1/2 and JNK, but not of p38 kinase in cultured human dermal fibroblasts (Fig. 2C). ERK and JNK phosphorylation peaked at 15 min post-MβCD treatment. However, the phosphorylation of p38 kinase tended to decrease with cholesterol depletion by MβCD (Fig. 2C). Using EMSA, we demonstrated that cholesterol depletion by MβCD treatment increased the DNA binding activity of AP-1 transcription factor. AP-1 binding activity was greatly increased at 60 min after MβCD treatment (Fig. 2D).
Then, we examined the effects of MAPK specific inhibitors, including MEK inhibitor (U0126), JNK inhibitor (SP600125), and p38 kinase inhibitor (SB203580) on MβCD-induced MMP-1 expression in cultured human dermal fibroblasts. Cells were pretreated with each inhibitor for 30 min and then further incubated with 10 mM MβCD for 1 h. Inhibition of MEK and JNK pathways by U0126 and SP600125, respectively, completely blocked MβCD-induced MMP-1 expression. Inhibition of p38 kinase by SB203580 did not affect the MβCD-induced MMP-1 expression significantly (Fig. 2E). These data suggest that MMP-1 induction by cholesterol depletion may be mediated by activation of ERK1/2- and JNK-dependent pathways in cultured human dermal fibroblasts. Our results indicate that cholesterol depletion increases the phosphorylation of ERK and JNK, which may lead to activation of transcription factor AP-1.
Cholesterol repletion decreased the cholesterol depletion-induced expression of MMP-1 mRNA and protein through inhibition of ERK/JNK and AP-1-dependent signaling in cultured human dermal fibroblasts
In the next study, we investigated the effects of cholesterol repletion on MβCD-induced expression of MMP-1 mRNA and protein in cultured human dermal fibroblasts. The cells were treated with 10 mM MβCD with or without 100µg/ml cholesterol for 1 h, and then the cells and culture media were harvested to measure the MMP-1 mRNA and protein expression, respectively. Cholesterol depletion by MβCD increased the level of MMP-1 mRNA by 580% of the control level, and repletion of 100µg/ml of cholesterol reduced the MβCD-induced MMP-1 mRNA expression to the control level (Fig. 3A). MβCD-induced MMP-1 protein expression also increased significantly by 450% of the control level, and cholesterol repletion (100 µg/ml) significantly inhibited the MβCD-induced MMP-1 protein expression to 50.5±10.2% of the MβCD-treated cells (Fig. 3B). Next, we investigated the effects of cholesterol treatment on MβCD-induced phosphorylation of ERK1/2 and JNK in cultured human dermal fibroblasts. Cholesterol treatment decreased MβCD-induced phosphorylation of ERK1/2 and JNK (Fig. 3C). We investigated the effects of cholesterol depletion and/or repletion on AP-1 DNA binding activity in cultured human dermal fibroblasts. As shown in Fig. 3D, cholesterol depletion by MβCD treatment significantly increased the DNA binding activity of AP-1 transcription factor. On the other hand, MβCD-induced AP-1 DNA binding activity was suppressed by cholesterol treatment (Fig. 3D). Therefore, we demonstrated that cellular cholesterol level may regulate MMP-1 expression via ERK/JNK/AP-1 signaling pathway in cultured human dermal fibroblasts.
Cellular cholesterol level by cholesterol depletion and/or repletion in cultured human dermal fibroblasts
To confirm whether cellular cholesterol content is reduced by MβCD treatment in cultured human dermal fibroblasts, the cells were treated with 10 mM MβCD and/or 100µg/ml cholesterol for 1 h, and then the cellular cholesterol levels were measured. The cells were stained with the cholesterol-binding agent, filipin, to observe the cholesterol content inside the cells. MβCD treatment decreased the cholesterol content, while cholesterol repletion inhibited the MβCD-induced decrease in cholesterol (Fig. 4A~C). The level of cellular cholesterol was reduced in the MβCD-treated cells by an average of 58.2±5.2%, compared with that in the control cells (Fig. 4D). Cholesterol treatment increased the cellular cholesterol level to 120.4±29.4% of the control cells (data not shown), and inhibited the decrease of cellular cholesterol in the MβCD-treated cells; the cholesterol level in the MβCD and cholesterol-treated cells was 87.5±6.2% of the control level (Fig. 4D).
DISCUSSION
Changes in cholesterol content, both by inhibition of its biosynthesis or by its removal from the plasma membrane, affect the localization of proteins associated with lipid rafts, and thus affecting their function26-28. However, the function of cholesterol is not fully understood during the skin aging and in particular, its function remains unclear in the regulation of MMP-1 expression which is an important in extracellular matrix (ECM) degradation and remodeling of the human skin. Therefore, this paper explored the effects of intracellular cholesterol on regulation of MMP-1 expression in cultured human dermal fibroblasts. We suggest that the intracellular cholesterol level regulates MMP-1 expression. Basal MMP-1 mRNA and protein expression was decreased by cholesterol treatment in cultured human dermal fibroblasts, while cholesterol depletion up-regulated MMP-1 expression.
Intracellular cholesterol activates various signaling pathways. Incubation with water-soluble cholesterol decreased EGF-induced EGFR tyrosine phosphorylation, suggesting that the presence of cholesterol negatively regulates EGFR kinase activity10. Moreover, cholesterol depletion increased the intrinsic tyrosine kinase activity of EGFR in membranes generated from MβCD-treated NIH3T3 cells6 and had a striking effect on tyrosine phosphorylation of endogenous cellular proteins such as EGFR and ERK29. It also was reported that acute depletion of membrane cholesterol increases phosphorylation of ERK29,30 and one of the cholesterol binding proteins, oxysterol-binding protein as found to function as a cholesterol-binding scaffolding protein coordinating the activity of phosphatases such as PP2A and HePTP to control the ERK signaling pathway30.
We also demonstrated that cholesterol depletion by MβCD activated ERK1/2 and JNK, increased the c-Jun phosphorylation and stimulated AP-1 DNA binding activity. The MβCD-induced phosphorylation of ERK1/2 and JNK was inhibited by cholesterol repletion. Previous studies have reported that MMP-1 induction is mediated by ERK1/2 and JNK activation in normal human skin fibroblasts31,32. Activation of ERK1/2 in human gingival and dermal fibroblasts induces MMP-1 and stromelysin-1 (MMP-3) expression33. Other reports have shown that p38α mediates activation of protein phosphatases 1 and 2A and blocks the ERK1/2 cascade at the level of MEK1/2, resulting in suppression of MMP-1 promoter activity34. TNF alpha-induced and heat-induced MMP-1 expression is mediated through ERK1/2 and JNK activation15,35. Consistent with these results, we demonstrated that cholesterol depletion by MβCD treatment induced MMP-1 expression and this increased MMP-1 expression was decreased significantly by MEK inhibitor, U0126, and JNK inhibitor, SP600125, suggesting that ERK1/2- and JNK-dependent pathways mediate MMP-1 expression in MβCD-treated fibroblasts.
It is well known that MMP-1 expression is found to be increased in aged and photoaged elderly human skin25. This increased level of MMP-1 in dermal fibroblasts is known to play a role in degrading the extracellular matrix in the dermis in UV-irradiated and aged human skin, leading to wrinkle formation and aged appearance31,32.
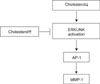
In summary, our data demonstrates that the intracellular cholesterol level modulates downstream signaling molecules including ERK1/2 and JNK, and then regulates MMP-1 mRNA and protein levels (Fig. 5). Based on these results, we suggest that cholesterol is an important negative regulator of MMP-1 expression in human dermal fibroblasts. Therefore, increasing cholesterol levels may provide a good strategy to prevent MMP-1-mediated degradation of ECM in human skin.




 XML Download
XML Download