

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Both a defective epidermal permeability barrier1-4, and a propensity to develop secondary infections5 are well-recognized features of atopic dermatitis (AD). While it has been previously assumed that these abnormalities reflect Th2-driven immunologic abnormalities (the historical 'inside-to-outside' view of AD pathogenesis), we and others have long proposed that the barrier abnormality is not merely a secondary phenomenon, but rather the 'driver' of disease activity in AD ('outside-to-inside' view of disease pathogenesis)6-8, because it was well-known that: 1) the extent of the permeability barrier abnormality parallels severity of disease phenotype in AD1,2,4; 2) clinically-uninvolved skin sites, as well as skin cleared of inflammation for ≤5 years, continue to display barrier abnormalities2; 3) emollient therapy comprises effective ancillary therapy for AD9. Finally, 4) as will be discussed further below, specific lipid replacement therapy, which targets the prominent lipid abnormalities that account for the barrier abnormality6,9, not only corrects the barrier abnormality, but also ameliorates inflammation in AD.
The outer epidermis generates a set of protective and sensory functions, in part attributable largely to its anucleate, but metabolically-active, differentiation end-product, the stratum corneum (SC)10. These defensive functions include: 1) the permeability barrier, which both retards transcutaneous evaporative water loss in a potentially-desiccating external environment; 2) an antimicrobial barrier, which simultaneously encourages colonization by non-pathogenic 'normal' flora, while resisting growth and invasion by microbial pathogens11; 3) multiple other, key protective functions, such as antioxidant and defense against ultraviolet irradiations. Finally, 4) recently-appreciated, biosensory functions clearly place the epidermis as the distal outpost of the nervous system12, as well as performing important regulatory and signaling functions within the epidermis itself.
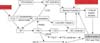
The SC comprises a multilayered tissue composed of vertically-stacked arrays of anucleate corneocytes, embedded in a hydrophobic extracellular matrix filled with multilayers of planar lamellar bilayers, enriched in a family of at least 10 ceramides (Cer), cholesterol, and both essential and non-essential free fatty acids (FFA). These lipids are delivered to the SC interstices as their precursors (i.e., glucocylceramides, sphingomyelin, cholesterol sulfate, and phospholipids) through secretion of the epidermal lamellar body (LB) contents (Fig. 1). However, this epidermis-unique organelle delivers not only lipid precursors, but also lipid hydrolases (β-glucocerebrosidase, acidic sphingomylinase, secretory phospholipase A2, and steroid sulfate), that generate Cer, cholesterol, and FFA, which then self-organize into lamellar membranes. In addition, LB-derived proteases/anti-proteases orchestrate the orderly digestion of epidermis-unique, junctional structures, corneodesmosomes (CD), allowing invisible shedding of corneocytes from the skin surface13,14. Pertinent to distal innate immunity, certain antimicrobial peptides (AMP) (i.e., the carboxy terminal fragment of hCAP18 [LL-37] and human β-defensin 2) also are delivered to the SC intercellular domains through secretion of LB contents15,16.
RELATIONSHIP OF ICHTHYOSIS VULGARIS TO ATOPIC DERMATITIS
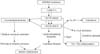
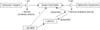
Preliminary studies in genotyped AD subjects, and in filaggrin (FLG)-deficient mouse models17,18 suggest that the phenotype in both ichthyosis vulgaris (IV) and AD is linked to an underlying abnormality in permeability barrier homeostasis19. Moreover, as in AD, the pH of SC is elevated in IV19,20, and the increase in pH, in turn, could activate serine proteases (kallikreins), with a host of negative downstream consequences, including: a) plasminogen activator type 2 receptor (PAR2)-mediated blockade of LB secretion21,22; b) possible downstream alterations in keratin filament organization that could impede LB secretion (see below); and c) both Th1- and KLK5-activated Th2 inflammation23 (Figs. 2, 3). According to this view, while the primary phenotype in IV is one of scaling, it also represents the forme fruste of AD, displaying clinical inflammation only when affected skin is either exposed to sustained antigen ingress, and/or to additional acquired stressors to the barrier (e.g., high pH surfactants, exposure to a reduced external humidity, or sustained psychological stress).
The strongest evidence that a primary structural abnormality of SC underlies the pathogenesis of AD derives from the recent link between loss-of-function mutations in the gene encoding, filament aggregating protein (FLG), and AD24,25. Up to 60% of Europeans with AD reveal single or double allele mutations in FLG on chromosome 1q21. FLG is the main component of F-type keratohyalin granules, responsible for the designation of the stratum granulosum. Decreased FLG expression results in a paucity of keratohyalin granules, a hallmark of IV26 and reduced FLG is also common in AD3,27,28. Accordingly, IV is associated with concomitant AD, allergic rhinitis and/or asthma in approximately two thirds of patients3.
FLG deficiency has been ascribed to both nonsense and frameshift mutations. Although more than 20 different mutations have been reported, 6 of them account for the majority of European cases29,30. Most FLG mutations result in truncation of pro-FLG, explaining reduced-to-absent FLG expression in the epidermis of IV/AD. While heterozygous patients show residual FLG with a milder phenotype, IV patients with homozygous or compound heterozygous mutations lack FLG, and exhibit generalized scaling, as well as an increased propensity to develop severe and persistent AD (op. cit.).
The initial product of FLG translation is pro-FLG, a large, histidine-rich, highly cationic phosphoprotein, consisting of 10~12 FLG repeats, enriched in hydrophobic amino acids31-33. Pro-FLG contains an amino-terminal sequence, including a calcium-binding A domain; the B domain is a putative S100-like, calcium binding domain. In contrast to the cytoplasmatic localization of C-terminal FLG monomers, the N-terminus of pro-FLG appears to tether to the nucleus via its nuclear localization sequence. In normals, pro-FLG is dephosphorylated and proteolytically processed to FLG monomers during cornification. Processed FLG peptides then induce aggregation of keratins within the corneocyte cytosol, and attach to the cornified envelope (CE), a unique structure that replaces the plasma membrane as granular cells transform into corneocytes34,35. The CE provides a relatively-inflexible, mechanically-resistant barrier. However, as the water content of the SC drops in the mid-to-outer SC, FLG detaches from the CE, and the C-terminal portion of FLG is proteolyzed into its constituent amino acids, followed by their deimination into polycarboxylic acids (=natural moisturizing factors, NMF)36,37, such as pyrrolidine carboxylic acid and trans-urocanic acid (t-UCA)38. These metabolites, in turn, act as osmolytes, drawing water into corneocytes, thereby accounting in large part for corneocyte hydration.
While it is widely hypothesized that FLG deficiency provokes a permeability barrier abnormality25, until very recently, the cellular basis for such an abnormality is unknown. Indeed, abnormal permeability barrier function was noted in IV patients, lacking AD, in the era prior to genetic FLG analysis39-41. But Hubiche et al., (2007) failed to find defective barrier function in IV42, thereby challenging the prevailing hypothesis that FLG deficiency causes an impaired barrier to transcutaneous water loss. Our recent studies confirm that both single- and double-allele IV patients display a barrier abnormality43. Yet, how loss of FLG (an intracellular protein) provokes a permeability barrier abnormality (almost always an extracellular defect) is only now becoming clear (Figs. 2, 3). One possibility is that loss of this quantitatively-important protein could alter corneocyte shape, perhaps inducing flattening that could disrupt extracellular lamellar bilayer organization. Alternatively, our recent results suggest that decreased FLG cornified envelope integrity, contributing to poor SC integrity. Moreover, our very recent studies suggest yet another mechanism; i.e., that unprocessed pro-FLG interferes with loading of LB contents, as well as secretion44. But the most immediate result of FLG deficiency in AD likely is decreased SC hydration, which leads to a steeper water gradient across the SC, thereby 'driving' increased transcutaneous water loss. Thus, decreased SC hydration, leading to increased water loss, is likely an important cause of barrier dysfunction in FLG-deficient AD.
Yet, none of these mechanisms alone suffice to explain enhanced antigen penetration in AD, which could best explained by yet another consequence of FLG deficiency; i.e., decreased downstream production of acidic metabolites resulting from FLG proteolysis (Fig. 2). Indeed, t-UCA, in particular, is a purported, endogenous acidifier of the SC45. Thus, decreased generation of FLG products could result in an initial increase in the SC pH, sufficient activate multiple serine protease (SP) in SC, which all exhibit neutral-to-alkaline pH optima14. Such a pH-induced increase in SP activity, if prolonged, could precipitate multiple downstream structural and functional alterations, including Th2 inflammation in AD (see below).
LINK BETWEEN NETHERTON SYNDROME AND ATOPIC DERMATITIS
Netherton syndrome (NS) always presents with concurrent, severe AD, including elevated IgE levels, mucosal atopy, anaphylactic food reactions, and a characteristic hairshaft abnormality, trichorrhexis invaginata ('bamboo hair'). Because of the severity of AD, an exploration of pathogenic mechanisms in NS could provide further insights into AD pathogenesis (Fig. 3). NS is associated with loss-of-function mutations in SPINK5, which encodes the SP inhibitor, lymphoepithelial Kazal-type inhibitor 1 (LEKTI 1)46. Diminished LEKTI 1 activity in NS likely leads to cutaneous inflammation by multiple mechanisms. First, because SPs activate IL-1α/β in corneocytes47, initiating the 'cytokine cascade'48; and second, an acquired deficiency of LEKTI 1, due to proteolytic consumption of the inhibitor, could also contribute to the pathogenesis of AD49 (Fig. 4). Third, KLK5 can activate Th2 cytokines, independent of allergen exposure23. Moreover, in some population studies, polymorphisms in the SPINK5 gene (missense variants) also are associated with AD and mucosal atopy50. Yet several control studies have been found with an increased frequency of single nucleotide polymorphisms (Glu420Lys) in SPINK550, recent studies cast doubts upon this association. Likewise, a British case-control study describing putative, gain-of-function polymorphisms (AACCAACC vs. AACC) in the 3' region of KLK7, which encodes the SP, SC chymotryptic enzyme (KLK7)51, is now disputed. Furthermore, in a recent genetic study involving 2,500 AD cases and 10,000 controls, there was no evidence for an epistatic (additive) interaction between SPINK5/KLK7 polymorphisms and FLG mutations52. Yet, transgenic mice that express human KLK7 display a severe AD-like dermatosis. Moreover, in experimental animals, a net increase in SP activity, achieved by a variety of means, has been shown to compromise barrier function through accelerated degradation of both CD (accounting for flawed SC integrity) and lipid processing enzymes53 (Fig. 2), resulting in a failure to generate Cer, a characteristic lipid abnormality in AD54,55. Furthermore, residual LEKTI expression in NS correlates inversely with the extent of SP activation within the outer epidermis56, resulting in a gene dose-dependent permeability barrier defect56, due to unrestricted, SP-dependent degradation of CD and Cer-generating hydrolases56.
In addition to KLK5 upregulation of Th2 inflammation via signaling of the PAR2, elevated SP activity also can provoke a barrier abnormality via PAR2-induced down-regulation of LB secretion57, entombing some of these organelles in nascent corneocytes58. Partial failure of LB secretion accounts, in turn, for the global decrease in SC lipids in AD3,59, which correlates with a decrease in extracellular lamellar bilayers in AD4. Thus, increased SP activity provides a mechanistic basis for the immunologic and lipid abnormalities, as well as the further decline in Cer levels that occur in AD (Fig. 2).
FLAWED ANTIMICROBIAL DEFENSE IN ATOPIC DERMATITIS
The antimicrobial barrier also is compromised in AD, commonly leading to colonization of lesional and non-lesional skin by Staphylococcus aureus5, initiating yet another series of vicious cycle in AD. Although colonization is most-often not apparent, impetiginization, widespread folliculitis, or less-frequently, cutaneous abscesses or cellulitis, are well-recognized complications in AD. Elevated SP activity can degrade AMP, particularly LL-37, into smaller fragments, which lack antimicrobial activity. Moreover, superantigen producing S. aureus strains colonize AD more commonly in steroid-resistant patients60, and further exacerbate disease in AD through augmentation of IgE production, as well as through stimulation of IgE, specifically directed towards epidermal structural proteins (rev. in19). Over time, non-toxigenic strains of S. aureus that colonize AD can be replaced by enterotoxin-generating strains61, which in turn, could aggravate AD by at least 3 mechanisms: 1) toxigenic strains are more likely to produce clinical infections than are non-toxigenic strains61; 2) some toxins stimulate pruritus62 and production of specific IgE5,63; and 3) some toxins serve as 'superantigens' that stimulate T and B cell proliferation, as well as immunoglobulin class-switching to allergen-specific or 'superallergens' that stimulate IgE production5. Activated T cells produce IL-31, which also induces pruritus64. Finally, clinical infections, particularly folliculitis, are notoriously pruritic, even in non-atopics, eliciting an 'itch-scratch' vicious cycle that creates additional portals of entry for pathogens. It is self-evident that excoriations create further defects in the permeability barrier, representing yet another potentially-important vicious cycle in AD pathogenesis. Finally, patients with AD are also susceptible to widespread cutaneous viral infections, including molluscum contagiosum, Herpes simplex (Kaposi's varicelliform eruption), and life-threatening Vaccinia infections. Widespread dermatophytosis (tinea corporis) and Malassezia infections also occur in AD, and the latter, can also stimulate specific IgE production. Together, these observations point to loss of a competent antimicrobial barrier in AD. While failure of both permeability and antimicrobial function is well-recognized in AD, only recently has it become clear that these 2 functions share common structural and biochemical features11, and both are co-regulated and interdependent65 (Table 1). Thus, failure of the permeability barrier in itself favors secondary infection; and conversely, pathogen colonization/infection further aggravates the permeability barrier abnormality.
Increased colonization with S. aureus66 occurs both as a result of the barrier abnormality (a structurally-competent, lipid-replete, acidic SC itself comprises a formidable barrier to pathogen colonization11, and it can further aggravate barrier function in AD by several mechanisms. The antimicrobial barrier is intimately linked to the permeability barrier65, and as with water egress, pathogen ingress occurs via the extracellular domains67. Moreover, an impaired permeability barrier alone predisposes to pathogen colonization, not only because of the increase in surface pH, but also because levels of FFA and the Cer metabolite, sphingosine, which exhibit potent in vitro antimicrobial activity67,68, decline in AD11. Surface proteins on S. aureus can down-regulate epidermal FFA production, thereby aggravating both permeability and antimicrobial function in parallel, a strategy that could also facilitate microbial invasion. In addition, at least one member of epidermal AMP, the human cathelicidin product, LL-37, is down-regulated in a Th2-dependent fashion in AD66,69, and LL-37 is required for normal permeability barrier function65. Notably, LL-37 also displays robust activity against S. aureus, and is required not only for normal epidermal permeability barrier function, but also for the integrity of extracutaneous epithelia. Thus, it is likely that decreased LL-37 amplifies the barrier defect in AD.
EXOGENOUS AND ENDOGENOUS STRESSORS AGGRAVATE BARRIER FUNCTION IN AD
That FLG mutations alone do not suffice is shown in IV, where the same single or double allele FLG mutations reduce FLG content, but inflammation (i.e., AD) does not inevitably occur. We and others have repeatedly proposed that certain stressors aggravate the barrier abnormality70-72. One or more of these stressors likely provoke an incremental increase in pH of the SC, leading to a further amplification of SP activity (Fig. 2). For example, pH-dependent increases in SP activity likely accounts for the precipitation of AD following the use of neutral-to-alkaline soaps73.
Prolonged exposure to a reduced environmental humidity, as occurs in radiant-heated homes in temperate climates during the winter, is also a well-known risk factor for AD. Under these conditions, transcutaneous water loss would accelerate across a defective SC, aggravating the underlying hydration and permeability barrier abnormalities, and amplifying cytokine signaling of inflammation. Because FLG proteolysis is regulated by changes in external humidity38, sustained reductions in environmental relative humidity could further deplete residual FLG in single-allele FLG-deficient patients. Finally, sustained psychological stress aggravates permeability barrier function in humans74, and PS is both a well-known precipitant of AD, and cause of resistance to therapy. In experimental animals, PS induces an increase in endogenous glucocorticoids (GC), which in turn alter permeability barrier homeostasis, SC integrity and epidermal antimicrobial defense70,71. The putative mechanism for the negative effects of PS is GC-mediated inhibition of synthesis of the three key epidermal lipids that mediate barrier function; i.e., Cer, cholesterol, and FFA. Accordingly, a topical mixture of these three lipids largely normalizes all of these functions, even in the face of ongoing PS or GC therapy75,76, and should comprise particularly effective therapy for AD patients with unusual levels of stress.
BASIS FOR INFLAMMATION IN AD
As noted above, one important downstream determinant of cutaneous inflammation in AD results from increased SP activity, which activates the primary cytokines, IL-1α and IL-1β47, from their 33 kDa pro-forms that are stored in large quantities in the cytosol of corneocytes. The putative pH-induced increase in SP activity would generate 17 kDa active forms of these cytokines47, the first step in the cytokine cascade that we have long proposed is an important contributor to inflammation in AD6,7. Sustained antigen ingress through a defective barrier leading to a Th2-dominant infiltrate then would be a second cause of inflammation in AD70 (Fig. 3). Certain antigens, such as cat dander, mites, and cockroach antigens, are preferentially associated with AD and are frequent triggers of AD, particularly in FLG-deficient patients77. Mites themselves activate SP activity with further damage to the barrier78. Yet, the damaged barrier in AD is due to lipid depletion, which also explains the preferential penetration of water-soluble haptens, such as nickel, in AD79. Accordingly, correction of the barrier abnormality alone should ameliorate both the cytokine cascade and allergen-induced inflammation in AD.
Despite accumulating evidence in support of a barrier-initiated pathogenesis of AD, recent studies suggest further mechanisms whereby Th2-generated cytokines could also further aggravate AD70. Exogenous application of the Th2 cytokines, IL-4 or IL-13, impede permeability barrier homeostasis in experimental animals80. The basis for these negative effects of IL-4 includes: 1) inhibition of ceramide (Cer) synthesis81, providing yet another mechanism accounting for decreased Cer in AD; 2) inhibition of keratinocyte differentiation-linked proteins, most notably loricrin and FLG27; and 3) decreased desmoglein 3 expression, which would further compromise SC integrity80,81. In a further 'vicious cycle,' serum IgE from AD patients auto-reacts against a variety of keratinocyte antigens82. Together, these observations provide acquired mechanisms that could further compromise barrier function in AD27. Thus, primary inherited barrier abnormalities in AD ultimately stimulate downstream paracrine mechanisms that further compromise permeability barrier function, completing a potential 'outside-inside-outside' pathogenic loop in AD (Fig. 2).
THERAPEUTIC IMPLICATIONS
Anti-inflammatory steroids and immunomodulators remain the mainstay of therapy in AD. Yet, the evidence cited above creates a powerful rationale that measures aimed at reducing the pH of SC, reducing SP activity, and/or corrective lipid replacement could prevent and/or ameliorate the inflammatory disease component in AD, and could break the vicious cycle of inflammation-induced barrier impairment27. We provide below some reasons for concern with over-reliance on these agents.
Even though anti-inflammatory therapies effectively reduce disease severity by suppressing immune function in AD, they do not address the primary underlying barrier abnormality that 'drives' disease pathogenesis70. Reductions in inflammation alone can, however, reduce TEWL due to the 'vicious cycle' that is operative in AD. As noted above, recent studies have shown that topical immunomodulators compromise barrier function in normal skin83, as do topical glucocorticoids76,84. Thus, as AD improves, continued topical applications of steroids and immunosuppressants (IS) likely begin to compromise barrier function. In contrast, studies in both AD animal models and in patients show that corrective lipid replacement therapy reduces the inflammatory component of disease by promoting normal epidermal function70,72,85,86.
Because of the numerous other protective functions of the outermost skin layer (SC), the rationale for barrier repair therapy goes beyond trapping moisture and preventing dry skin. The 'pro-inflammatory' functions of the SC begin with the large pool of pre-formed IL-1α and IL-1β, which are stored in large quantities in corneocytes. These molecules are released into the lower epidermis and dermis when barrier function is perturbed, helping to normalize function in normal skin. Since barrier is always abnormal in AD, these repair mechanisms are unsuccessful in AD. Hence, the cytokine cascade is sustained, resulting in the downstream recruitment of additional, pro-inflammatory molecules (the 'outside-to-inside' concept of AD pathogenesis). In addition to the ongoing cytokine cascade, repeated access of haptens across a defective barrier ultimately stimulates the characteristic TH2-cytokine response70,71, and SP activation alone can initiate Th2 inflammation23. Along with exotoxins released from colonizing S. aureus, these 4 barrier-initiated mechanisms account for downstream inflammation in AD. Together, these data provide a strong scientific rationale for barrier repair therapy in AD.
Although emollient moisturizers decrease steroid use through moisturization87, they consist of non-physiologic lipids, such as petrolatum and lanolin, which actually impede, rather than correct the underlying biochemical response to a defective in barrier in AD88. Yet, effective, disease-specific formulations that correct the underlying barrier abnormality have only become available recently. The permeability barrier defect in AD is characterized by a global reduction in the contents of all three key lipids (i.e., cholesterol, FFA, and Cer), with a further reduction in ceramide content54,55. Thus, correction of the barrier abnormality in AD requires topical applications not only of sufficient quantities of all three key lipids that mediate barrier function, but also provision of the lipids in a ceramide-dominant proportion that corrects the underlying lipid biochemical abnormality in AD88. Under these conditions, restoration of normal skin barrier function then can down-regulate inflammation in deeper skin layers.
Several recent studies show that targeted correction of the lipid biochemical abnormality in AD, with a ceramide-dominant form of barrier repair therapy (EpiCeram® emulsion, (Promius Pharma, LLC, Bridgewater, NJ, USA)), comprises effective therapy for AD85. Since EpiCeram® is formulated at an acidic pH, and only contains ingredients that are present (individually) in currently-available moisturizers, it displays none of the safety concerns of either topical steroids or IS, and therefore it could assume a central place in the treatment of AD85. Whether barrier repair therapy will also reduce secondary colonization by pathogenic S. aureus, and whether it will prevent emergence of mucosal atopy remains to be determined. Notably, several other currently-available products that make 'barrier repair' claims incorporate either incomplete mixtures of the 3 key lipids, incorrectly formulated mixtures, incorrect types of the three lipids, or in some cases, insufficient quantities of the three lipids85. Not surprisingly, these products fail to improve barrier repair in animal models of AD (op. cit.). The defective epidermal barrier in AD could be that it would allow epicutaneous delivery of antigens that induce asthma and allergic rhinitis. Thus, the 'atopic march'; i.e., the tendency for AD to precede the later development of mucosal atopy, can be explained by cutaneous penetration of aeroallergens of all types. FLG deficiency is associated with mucosal atopy, independent of AD89, though FLG is not expressed in either bronchial or other non-keratinizing mucosal epithelia90. Thus, it is likely that lipid replacement therapy could block development of the 'atopic march'.


 XML Download
XML Download