

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Incontinentia pigmenti (IP, Bloch-Sulzberger syndrome), first reported by Bloch1 in 1926 and Sulzberger2 in 1928, affects the skin in three or four stages: vesicular, verrucous, and hyperpigmented, with or without an atrophic stage. The skin lesions typically follow the lines of Blaschko. IP is frequently accompanied by abnormalities of the teeth, eyes, nervous tissue, hair, nails, musculoskeletal system, and heart3,4 and may be associated with immunologic derangement5. We describe a patient with IP associated with a ventricular septal defect (VSD), left hemiatrophy, hemangiomas, an abnormal labial frenum, and spastic cerebral palsy manifested as left hemiplegia and developmental delay.
CASE REPORT
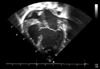
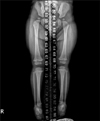
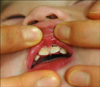
A 6-month-old girl born at 39-weeks gestation and weighing 3,100 g was referred to us with linear and whorled brownish patches on her trunk and extremities (Fig. 1A, B). At birth, two reddish plaques, thought to be hemangiomas, were found on her occiput and back (Fig. 1C). At 1 week of age, she developed dyspnea. Echocardiography revealed a large perimembranous VSD (Fig. 2), for which she underwent VSD patch closure. When she was about 10 days old, erythematous and vesicular lesions developed on her extremities. At 2 months of age, linear and whorled brownish patches appeared on her trunk and extremities in the distribution of Blaschko's lines. These patches remained until she was 6 months old, at which time a punch biopsy was taken. Histopathologic examination showed focal vacuolar changes of the basal layer, as well as a few eosinophils and dermal melanophages (Fig. 3). When the patient was 6 months old, a discrepancy in limb length was noted, with her right leg 1.4 cm longer than her left leg at 14 months (Fig. 4). At 8 months, a rehabilitation specialist diagnosed her with spastic cerebral palsy, manifested as left hemiplegia and developmental delay; she has been in physical therapy ever since that time. At 11 months, she developed an abnormal labial frenum (Fig. 5), but she had no dental abnormalities. No other person in her family has a similar disorder. Her mother denied any harmful exposures during pregnancy, such as drugs, alcohol, smoking, or radiation. Prenatal examination revealed no chromosomal abnormalities or signs of VSD. Although our patient's pediatrician recommended a molecular analysis, her parents were unable to do so due to economic concerns.
DISCUSSION
IP is a rare genodermatosis, inherited in an X-linked dominant fashion. It is lethal in males, resulting in an observed female-male ratio of 37:14. IP has been observed, albeit rarely, in males with somatic mosaicism, Klinefelter syndrome (47, XXY), and hypomorphic alleles6. IP is caused by mutations in the NEMO/IKKγ gene located on Xq287. The most common NEMO abnormality in IP is a deletion of exons 4 through 10, which occurs in 70~80% of IP patients8,9. The remaining patients have other alterations in NEMO, including small duplications, substitutions, and deletions.
IP can affect ectoderm-derived structures, including the skin, teeth, eyes, nervous tissue, hair, and nails3,4 and may be seen in association with abnormalities of the musculoskeletal system and heart3,4. In a study of 653 IP patients, 521 (79.8%) had systemic symptoms and signs4.
Three or four stages of IP have been reported in the skin. In the vesicular stage (Stage I), erythema, vesicles, and pustules appear in a linear arrangement within the first 2 weeks of life; the extremities and trunk are the most affected areas. In the verrucous stage (Stage II), hyperkeratotic verrucous papules and plaques appear at about 2 months of age. The hyperpigmented stage (Stage III), which is characterized by irregularly whorled hyperpigmented patches, follows as the verrucous lesions resolve. In some patients, an atrophic stage (Stage IV) occurs. Cutaneous lesions in all stages tend to follow Blaschko's lines10. Histopathologically, stage I is characterized by eosinophilic spongiosis, intraepidermal vesicles, and dermal infiltration, and the number of eosinophils may increase in the peripheral blood. Stage II is characterized by dyskeratotic keratinocytes, hyperkeratosis, acanthosis, and papillomatosis, and stage III is characterized by vacuolar changes of the basal layer and several dermal melanophages. In stage IV, skin appendages may be absent, with mild epidermal atrophy and decreased, normal, or small melanocytes.
Scarring alopecia has been observed in 28% to 38% of IP patients, most commonly on the vertex11. Forty percent of IP patients have nail abnormalities12. Onychogryphosis, pitting, and yellow discoloration have been reported, as have benign subungual dyskeratotic tumors3,4; a port-wine stain has been reported in one patient with IP12.
Between 65% and 80% of IP patients have dental abnormalities3, such as anodontia, hypodontia, delayed dentition, impaction, pegged teeth, conical crown formation, and accessory cusps3,4,11,12. In addition, 35% to 40% of IP patients have ocular abnormalities3, most commonly strabismus, but also including cataracts, keratitis, and optic nerve atrophy3,4,11,12. Avascularity in the peripheral temporal retina, retinal detachment, and preretinal fibrovascular proliferation with vitreous hemorrhage have also been observed, all of which may lead to blindness3,4,11,12.
Between 10% and 31% of IP patients have neurologic symptoms and signs3. Seizures are the most common symptoms, but mental retardation, microcephaly, spastic or paralytic quadriplegia, hemiplegia, and diplegia have also been reported in IP patients3,4,11,12. Abnormalities of the musculoskeletal system have been observed, including hemivertebra, hemiatrophy, syndactyly, congenital dislocation of the hip, club foot, dwarfism, kyphoscoliosis, unilateral acheiria, and supernumerary ribs3. Among the cardiovascular anomalies observed in patients with IP are atrial septal defects, patent ductus arteriosus, acyanotic tetralogy of Fallot (TOF), ventricular endomyocardial fibrosis, tricuspid insufficiency, and primary pulmonary hypertension3,4,11,13. Cleft lip and palate have been rarely reported14. Immunologic abnormalities are common in IP5 and include functional abnormalities of neutrophils and lymphocytes and defects in polymorphonuclear chemotaxis.
At her first visit, our patient presented with characteristic whorled brownish patches on her trunk and extremities, and histopathologic findings were consistent with stage III IP. She also had a large perimembranous VSD, left hemiatrophy, hemangiomas, an abnormal labial frenum, and spastic cerebral palsy manifested as left hemiplegia and developmental delay. To our knowledge, although VSD is a relatively common cardiac anomaly estimated to affect up to 1% of babies, there has been only one reported case of an IP patient with TOF, one component of which is a VSD13. An abnormal labial frenum has also never been reported in an IP patient, and a cutaneous vascular anomaly has been described in only one case of IP, in which a port-wine stain was present. Spastic cerebral palsy with hemiplegia and developmental delay has also been rarely reported in patients with IP, as has hemiatrophy, manifested as a discrepancy in limb length. To date, our patient has shown no dental or ocular abnormalities.
Because IP is a systemic disorder, a multidisciplinary approach to management is crucial. Our patient underwent VSD patch closure under the care of a pediatric cardiac surgeon, and the hemangioma on her back was treated by a dermatologist using a V-beam laser. She is also seeing a rehabilitation specialist for physical therapy of her spastic cerebral palsy. In the future, labial frenectomy should be performed by a dental surgeon. An orthopedic treatment such as the Ilizarov technique may be necessary to balance the length of her limbs. Since she is only 11 months old, careful follow-up examinations by an ophthalmologist and a dentist will be necessary in the upcoming months and years.


 XML Download
XML Download