

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Dystrophic epidermolysis bullosa (DEB) is a group of heritable mechanobullous disorders that's characterized by blistering and scarring of the skin and mucosae and nail dystrophy that's all induced by minor trauma1,2. The ultrastructural hallmark of DEB is tissue separation at the sublamina densa level, and this condition is usually associated with an abnormal quality or quantity of anchoring fibrils at the dermal-epidermal junction1,3,4. Dystrophic epidermolysis bullosa is inherited either in an autosomal dominant or recessive mode1,4. Recessive DEB (RDEB) is a much more serious form than dominant DEB, with the blistering generally being present from birth4. RDEB is classified into Hallopeau-Siemens and non-Hallopeau-Siemens2. We describe here a case of non-Hallopeau-Siemens RDEB that was diagnosed based on the family history and the clinical, histological and electron microscopic findings.
CASE REPORT
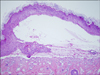
A 3.50 kg girl, who was delivered spontaneously at full term in January 2008 at a private hospital, was noted to have multiple bullae and denuded skin on the first day after birth over the areas where pressure and friction were applied. She was transferred to the neonatal intensive care unit of our hospital and she was referred to the Department of Dermatology for further evaluation and management. There were no other dysmorphic features and the girl was hemodynamically stable. There was no apparent history of skin diseases in her family. Her dermatologic examination revealed multiple serous or hemorrhagic blisters and skin abrasion on both hands, the feet and the scalp, and these blisters and abrasions subsequently extended to the trunk (Fig. 1). The laboratory studies, including a complete blood cell count with the differentials, the blood chemistry, VDRL, urinalysis, stool examination for occult blood and an infantogram were within nomal limits or they were negative. Wound swabs were positive for growth of Staphylococcus aureus and enterobacter species. The skin biopsy specimen from the right thigh showed subepidermal blisters (Fig. 2). Direct immunofluorescence of the perilesional skin from the upper arm demonstrated no immune deposits. Electron microscopy showed the lamina densa attached to epidermal keratinocytes at the roof of the cleft, and this was compatible with the diagnosis of DEB (Fig. 3). Based on the family history and the clinical, histological and electron microscopic findings, this case was diagnosed as non-Hallopeau-Siemens recessive dystrophic epidermolysis bullosa. She was treated with regular skin cleaning, dressings and 2% mupirocin ointment. She was also treated with phenytoin (7 mg/d), vitamin E (150 mg/d) and systemic antibiotics (amikacin 50 mg/d, vancomycin 240 mg/d) for 15 days, but no improvement was observed. She was scheduled to undergo genetic studies, but she was transferred to another tertiary hospital by her parent's wishes.
DISCUSSION
Epidermolysis bullosa (EB) is a complex group of inherited or acquired cutaneous diseases in which bullous lesions arise after normal levels of physical trauma. EB can be divided into three major general categories, based on the level of tissue separation within the cutaneous basement membrane zone that separates the epidermis from the dermis. In epidermolysis bullosa simplex (EBS), blisters occur within the epidermis. In junctional epidermolysis bullosa (JEB), the separation is in the lamina lucida of the dermoepidermal junction and in DEB, the lesions arise in the upper dermis1,4,5. The mode of inheritance is typically autosomal dominant in EBS and autosomal recessive in JEB. DEB can be either autosomal dominant or autosomal recessive6. RDEB is a much more serious form than dominant DEB, with blistering is generally present from birth4. RDEB is divided into Hallopeau-Siemens and non-Hallopeau-Siemens2. Hallopeau-Siemens RDEB is the most severe form of DEB and it is responsible for widespread mucocutaneous blistering leading to fusion of the digits, nail loss, flexural contractures, esophageal strictures, narrowing of trachea or larynx as well as oral and ocular erosions. Malnutrition, anemia and growth retardation commonly occur. Anal and genitourinary involvements may also be present. Squamous cell carcinoma is a common complication of the cutaneous scarring and it is a significant cause of mortality1,4. All the patients with RDEB who lack the cutaneous and extracutaneous features so characteristic of the Hallopeau-Siemens subtype are included under the term non-Hallopeau-Siemens RDEB2.
According to a National Epidermolysis Bullosa Registry report, 50 EB cases occur per 1 million live births. Of these cases, approximately 92% are EBS, 5% are DEB, 1% is JEB and 2% are unclassified. DEB occurs in all racial and ethnic groups and it equally affects males and females7. The epidemiologic data on this illness in Korea has been insufficient. In 1993, Rho et al8 surveyed 79 patients diagnosed with EB from 1970 to 1992 at the departments of dermatology and pediatrics in university hospitals and dermatologic departments in general hospitals in Korea. The epidemiologic study showed 8 cases of RDEB, and Kim et al9 reported 3 more cases in 2001. Yet because of the low incidence of EB in Korea, solid statistical data on this disease is still lacking.
The diagnosis of DEB is based on the clinical signs, the histopathology, the electron microscopy and, if available, genetic studies1,5,7. Separation of the epidermis and dermis was found on the histologic biopsy study of this case, and the electron microscopy showed the lamina densa attached to epidermal keratinocytes at the roof of the cleft, which is compatible with the diagnosis of DEB. Blisters and erosions were seen at birth, and this is also common in RDEB. The symptoms were generalized, but any extracutaneous features seen in the Hallopeau-Siemens subtype were not present, and the cutaneous features were mild. For those reasons, we were able to diagnose the patient as non-Hallopeau-Siemens RDEB.
Treatment for this disease is mainly symptomatic and supportive, as the currently available therapies cannot correct the underlying molecular defect. The treatment regimen is tailored to the severity and extent of the skin and systemic involvement and this usually entails a combination of wound management, infection control, surgical management as needed and nutritional support1,7. Administration of phenytoin has been experimented with for some recessive DEB cases for its inhibitory action on collagenase and it has been suggested as a therapeutic modality, but several studies have reportedly various responses9-12. Our patient was given phenytoin with vitamin E and systemic antibiotics, but she failed to show a significant response.
The symptoms of dominant DEB are mostly minor and it is possible to lead a normal life. But RDEB may lead to death that's caused by sepsis, depending on the degree of invasion, and the complication rates for such maladies as esophageal strictures and skin cancer are higher9,12. Therefore, a multidisciplinary approach is needed for these patients to assure an optimal outcome1,7. Especially, counseling and advise for the details of skin care must be provided and then reinforced to the parents of children who suffer with RDEB.


 XML Download
XML Download