

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Focal acral hyperkeratosis (FAH) was first described in 1983 by Dowd et al.1 as a rare disorder characterized by multiple, yellowish to white papules and plaques on the palms and soles, with preference for the palmar and plantar margins. Both sporadic and familial forms have been reported, with an apparent autosomal dominant inheritance in the familial cases. FAH is clinically similar to acrokeratoelastoidosis (AKE); both belong to the group of marginal papular acrokeratodermas (MPA)2. The histopathology, however, reveals subtle differences between the two; FAH does not have elastorrhexis. There has been only one previous report of FAH in a Korean patient; a 23-year-old female with a non-specific family history of FAH has been previously described3. We herein report a typical case of FAH in a 47-year-old Korean male with an autosomal dominant pattern of inheritance.
CASE REPORT
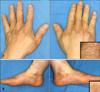
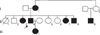
A 47-year-old male presented with multiple persistent flesh colored papules on the hands that were first noted during early adulthood. The number of lesions had gradually increased over the years. Physical examination revealed asymptomatic, multiple, firm, yellowish to flesh colored, hyperkeratotic polygonal papules clustered on the dorsum and lateral aspects of the hands and on the lateral borders of the feet (Fig. 1). The nails and hair were unremarkable. The patient had no history of local trauma, or exposure to arsenic. In addition, there was no history of atopy and no other significant medical history. The parents were non-consanguineous; he was the fourth of eight siblings, three of whom had similar skin lesions that developed during adolescence. His daughter, mother, and maternal cousin were also affected, with similar multiple flesh colored papules on the hands (Fig. 2).
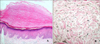
A skin biopsy specimen, taken from a representative lesion on the dorsum of the left hand, showed focal orthohyperkeratosis overlying a crateriform depression in a slightly acanthotic epidermis with slight hypergranulosis (Fig. 3A). No specific changes were detected in the dermis. The Verhoeff-van Gieson staining showed no evidence of elastorrhexis (Fig. 3B).
These clinical and histological findings were consistent with the diagnosis of FAH. Systemic treatment with oral acitretin 30 mg per day was initiated for the first two weeks, but the patient, after realizing the benign nature of the lesion, discontinued treatment.
DISCUSSION
FAH is a type of palmoplantar keratoderma (PPK), a heterogeneous group of disorders characterized by abnormal thickening of the palms and soles4. Rongioletti et al.2 further classified FAH as a type of MPA, in which the keratotic papules are distributed along the borders of the hands and feet. MPA includes several other disorders such as AKE, degenerative collagenous plaques of the hands, digital papular calcinosis, mosaic acral keratosis, and hereditary papulotranslucent acrokeratoderma.
Clinically, multiple flesh to yellowish colored papules and plaques are localized to the margins of the hands and feet. The lesions appear in the second or third decade of life, and gradually increase in number over several years. Both sporadic and familial forms have been reported, with an apparent autosomal dominant inheritance in the familial cases. For years, FAH was considered to be a racially limited focal disorder found in people of Afro-Caribbean or Arabic origin. However, cases have been reported in Caucasians2,5 and Asians3,6, suggesting that there is no racial preference.
Histologically FAH and AKE both show a pronounced orthohyperkeratosis overlying a crateriform depression in the epidermis. Hypergranulosis and mild acanthosis are commonly observed in both conditions. However, FAH lacks elastorrhexis, a major distinguishing feature between the two. Electron microscopic findings of FAH reveal normal collagen, elastic fibers, and fibroblasts1. The differential diagnosis of keratotic papules along the borders of the hands and feet that can be familial includes: acrokeratosis verruciformis of Hopf, degenerative collagenous plaques of the hands, digital papular calcinosis, mosaic acral keratosis, and hereditary papulotranslucent acrokeratoderma. They all exhibit similar clinical findings; however, the histological features aid in their differentiation. Acrokeratosis verruciformis of Hopf frequently exhibits papillomatosis and a frequent 'church spire' configuration, while degenerative collagenous plaques of the hands show thick collagen and elastic fibers, and digital papular calcinosis demonstrates elastosis, all of which are absent in FAH. The skin lesions in hereditary papulotranslucent acrokeratoderma are characteristically exacerbated after exposure to water.
The pathogenesis of FAH remains largely unknown. Many of the PPKs have underlying inherited genetic abnormalities for encoding the structural components of keratinocytes4, suggesting that a similar mechanism may underlie FAH. A recent report by Lee and Kim3 demonstrated increased expression of proliferation markers such as Ki-67 and PCNA in a patient with FAH, suggesting that the epidermal changes of FAH were caused by increased proliferation and differentiation of the keratinocytes in the lesion.
There is no effective treatment of FAH to date. Most patients are refractory to conservative treatment1,2,5. The treatment options for FAH aim to decrease the hyperkeratosis, such as the topical keratolytics including: urea, salicylic acid, and lactic acid, topical retinoids, and repeated physical debridement. Systemic treatment with acitretine has been considered as an effective alternative6. Treatment should conform to the patient's concerns since FAH is asymptomatic and benign, but potentially disfiguring.
FAH has been described infrequently in the English literature, and to our knowledge, there has been only one previous report of FAH in a Korean patient3. The case described a Korean female with FAH, but with no family history of a similar skin disorder. Our case depicts a typical presentation of FAH with associated family history in a Korean man. The diagnosis was based on the characteristic clinical and pathological findings. Currently, the origin of this condition remains unknown and the definition remains confusing and obscure. However, additional reports and molecular studies may aid in our improved understanding of FAH.
 XML Download
XML Download