

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Amelanotic malignant melanoma (AMM) is a subtype of cutaneous melanomas with little or no pigment on visual inspection. AMM constitutes 2~8% of all malignant melanomas, but the precise incidence is hard to evaluate because the term 'amelanotic' often indicates melanomas only partially devoid of pigment1. Four cases of AMM have been reported in the Korean dermatologic literature2-5.
Soft tissue sarcomas (STSs) are a heterogeneous group of neoplasms that arise from primitive mesenchymal cells. They are characterized by frequent somatic chromosomal rearrangements6. Liposarcoma is one of the most common STSs (15% to 23% of all STSs) occurring in adults6,7. Liposarcomas are generally deep-seated tumors most likely in the retroperitoneum or the deep soft tissues in or below the buttocks. They usually arise from intermuscular fascia, and only rarely from subcutaneous fat. Fletcher et al.8 classified liposarcomas into five major histopathological types. Well-differentiated sarcomas are more often encountered in older patients and have better prognoses than other subtypes (e.g., pleomorphic, round cell, and dedifferentiated types).
Previous studies have demonstrated multiple primary cancers in patients with STSs6,9,10. The coexistence of malignant melanoma and liposarcoma was also reported9,11. However, liposarcoma is uncommon in patients with melanoma9, despite the fact that it is the most common histologic subtype in all adult patients with sarcomas. Here we report a case of AMM coexisting with a well-differentiated liposarcoma.
CASE REPORT
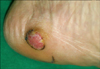
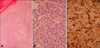
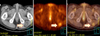
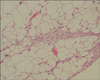
A 68-year-old male presented with a 5 month history of a slowly growing cutaneous lesion on the left heel without associated symptoms. Physical examination revealed a 1.5×1.5 cm well-demarcated eroded nodule with a dark-brown, hyperkeratotic border (Fig. 1). There was no inguinal lymph node enlargement. He had no remarkable past medical history and no family history of malignancy. Laboratory examinations were unremarkable. Under suspicion of eccrine poroma, clear cell hidradenoma, malignant melanoma, and other lesions, a punch biopsy was performed. A biopsy specimen showed numerous nests comprised of atypical epitheloid tumor cells with abundant mitoses in the dermis. The tumor cells had large hyperchromatic nuclei with prominent nucleoli and were arranged in solid nests which occupied the papillary and reticular dermis. These cells presented little melanin pigment on hematoxylin and eosin stain (Fig. 2A, B) and Fontana-Masson stain. On immunohistochemical study, the tumor cells were positive for S-100 (Fig. 2C) and HMB-45, but negative for cytokeratins. Because of these histologic findings, amelanotic melanoma was diagnosed. We performed whole-body PET/CT and bone scans for evaluating systemic involvement. PET/CT showed hypermetabolic lesions on the medial portion of the left sole (maximum SUV, 1.92) and gluteus maximus in the left buttock (maximum SUV, 1.22) (Fig. 3). Because of the diagnosis of amelanotic melanoma, he was transferred to the orthopedics department. Wide surgical mass excision of the tumor mass with sural artery flap and slit thickness skin graft were performed. The histologic evaluation of the excised specimen was consistent with the abovementioned findings. The lesion was Clark level IV, and the Breslow thickness was 5 mm. The PET/CT scan showed a 6×4×3 cm well-demarcated oval mass with increased uptake of fluorodeoxyglucose (FDG) in the left buttock. CT demonstrated low attenuation at the margin of the lesion and homogenous hyperdensity in the center. The lesion was widely excised and the excised specimen showed that the tumors were composed predominantly of univacuolated fat cells of various sizes. The nuclei of the fat cells were slightly pleomorphic, and some were hyperchromatic (Fig. 4). A fibrous septum containing cells with hyperchromatic nuclei was also observed. These histopathologic findings were consistent with a well-differentiated liposarcoma.
DISCUSSION
The synchronous or metachronous occurrence of two or more primary malignancies in one individual may have several etiologies12. It may represent an incidental occurrence, a result of host susceptibility factors (genetic predisposition or immunodeficiency), common carcinogenic influences from a clustering of different risk factors in the same individual, or it may be associated with treatment for the first tumor.
Many previous studies have demonstrated an increased incidence of other primary malignancies in patients with STSs6,9,10 such as fibrosarcoma, myxosarcoma, liposarcoma, leiomyosarcoma, synovial sarcoma, rhabdomyosarcoma, peripheral nerve sheath tumor, malignant fibrous histiocytoma, and dermatofibrosarcoma protuberans. A retrospective study between 1995 and 1999 demonstrated that of 375 patients with STSs, 28 (7.5%) developed other malignancies either before or after the diagnosis of STS10. Also, a large epidemiologic study in Sweden showed that a total of 650 (9.7%) second primary malignant neoplasms developed among 6671 first primary STS patients6.
An association between melanoma and sarcoma has been proposed by several investigators6,9,11. Berking and Brady9 reported 48 patients with melanoma and sarcoma. Among them, five patients (10%) presented with both tumors concurrently; 34 of 43 (79%) were diagnosed with melanoma first. The median interval between the two diagnoses was six years. Twenty-five percent of the patients had additional primary malignancies, and 50% of the patients had positive family histories of cancer, suggesting a predisposition for cancer in these patients. Only one patient (2%) in this study had a diagnosis of liposarcoma. Considering that liposarcoma accounts for approximately 23% (it is the most common histologic subtype) of all adult STSs in the general STS population7, this incidence is significantly low. The mechanism that explains this unusual finding is still unknown.
Numerous cytogenetic abnormalities have been implicated in melanoma as well as liposarcoma. The mutations in CDK4 or MDM2 may play a role in the pathogenesis of either. Germline and somatic mutations in the chromosome 9p21 tumor suppressor gene cyclin-dependent kinase inhibitor 2A (CDKN2A) are common and are a critical genetic event in the development of human melanoma13. CDKN2A engages the retinoblastoma (Rb) and p53 tumor suppressor pathways through its capacity to encode two gene products: p16 (also known as INK4a, inhibitor of kinase 4a) and p14ARF (alternative reading frame). p16 is a cell cycle regulator that binds and inhibits cyclin-dependent kinase CDK4, thereby inhibiting the progression of cells through the G1 phase of the cell cycle13,14. If p16 function is inactivated, unrestrained CDK4 activity phosphorylates the Rb protein, a cell-cycle regulator. This sequence culminates in enhanced cellular proliferation which results in neoplasms. Previous studies suggest that mutations in CDK4 result in a phenotype identical to that arising from p16 loss15. The p14ARF protein from CDKN2A inhibits a cellular oncogene, MDM2, which in turn promotes the demolition of the p53 tumor suppressor gene14. Thus, complete loss of CDKN2A also leads to the activation of MDM2 and loss of p53 function in melanoma.
Cytogenetically, well-differentiated liposarcomas are characterized by supernumerary circular "ring" chromosomes as well as giant long marker chromosomes composed of the 12q13-15 region and resulting in consistent amplification of MDM2 and CDK4 genes16. Hostein et al.17 reported CDK2 and MDM2 gene amplification in 82.4% and 98.2% of liposarcomas by real-time polymerase chain reaction (PCR), respectively.
The amplification of CDK4 and MDM2 genes may be related, providing a common pathogenesis in both melanoma and liposarcoma. However, further studies are required to determine whether these gene mutations play a key role in the coexistence of melanoma and liposarcoma. Furthermore, the studies of CDKN2A mutations were conducted in western countries13,14 and there is little information available on melanoma in non-white populations, especially genetic data18. Accordingly, it is not certain that CDKN2A mutations also lead to the same pathogenesis in Korean cutaneous melanoma patients. Although some authors have proposed that immunodeficiency may be related to the association of melanoma with STSs8, there is little evidence to support this relationship.
The patient in this report had no additional primary malignancies, family history for cancer, contact history to any carcinogens, or evidence of immunodeficiency. Also, he had never been exposed to radiotherapy or chemotherapy. The well-differentiated liposarcoma was found incidentally during evaluation for systemic involvement of AMM. His personal, medical, and familial history suggested that his case is an incidental occurrence of AMM and liposarcoma. However, to define whether mutations in CDK4 and MDM2 genes are related to this case, the evaluation of gene amplification by real-time PCR or immunohistochemical study will be required.
 XML Download
XML Download