

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Langerhans cell histiocytosis (LCH) is a rare, clinically polymorphous group of disorders all having in common proliferation of Langerhans cells. The recognized clinical variants of LCH include Letterer-Siwe disease, Hand-Schuller-Christian disease, eosinophilic granuloma, and congenital self-healing reticulohistiocytosis1. We report a 50-year-old man with brown lichenoid patches, unusual cutaneous presentation of LCH, on left dorsal foot.
CASE REPORT
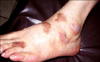
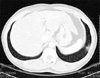
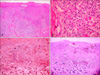
A 50-year-old man attended Department of Dermatology with a 2 year duration of brown-colored patches on the left dorsal foot. Lobular consolidation was shown on chest X-ray in health screening 5 years ago, but he had no symptoms. He was diagnosed pulmonary LCH by chest CT and percutaneous transthoracic needle aspiration. He is a heavy smoker and has 45 pack-years smoking history. On physical examination, there were three brown lichenoid patches on left dorsal foot (Fig. 1). Any regional lymph node is not palpable. His laboratory findings were all within normal range. Initial chest CT showed lobular consolidation at left basal segment of left lower lobe (Fig. 2). Regular enhanced chest CT in every 3 months revealed no interval change in pulmonary LCH lesion. A biopsy specimen of cutaneous lesion showed dense infiltrations in the dermis and dermoepidermal junction. The infiltrations consist of numerous histiocytes and few lymphocytes. Histiocytes appear as large, round cells with abundant cytoplasm and indented eccentric nucleus (Fig. 3A, B). Immunochemistry for CD 1a complex and S-100 protein showed positive staining but CD 68 show negative (Fig. 3C, D). The patient did not receive treatment but he visited our Department of Pulmonary Medicine regularly. To his last visit (1 year later) we can't find any changes on pulmonary and cutaneous lesions.
DISCUSSION
LCH encompasses a group of disorders of unknown origin with widely diverse clinical presentations and outcomes, characterized by infiltration of the involved tissues by large numbers of LC. LCH is characterized clinically by various combinations of systemic and cutaneous manifestations. LCH consists of Letterer-Siwe disease, Hand-Schuller-Christian disease, eosinophilic granuloma, and congenital self-healing reticulohistiocytosis1.
The definite diagnosis of LCH should fit light microscopy histology and at least 1 of the 2 following factors: 1) Birbeck's granules by electron microscopy, 2) labeling of CD1 antigen on pathological cells2. On light microscopy, the typical LCH cells are approximately four to five times larger than lymphocytes and have a vesiculated, reniform (kidney-shaped) nucleus and abundant, slightly eosinophilic cytoplasm. Sometimes, indeterminate cell histiocytosis (ICH) closely resembles LCH clinically and histologically. The cells in ICH are positive for S-100 protein, but show variable reactivity for CD1a. Macrophage markers, such as KP1 (CD68) and Ki-M1p, are often positive. No Birbeck granules are seen ultrastructurally3. In this case, we confirmed LCH by his past history, many LC infiltration in H&E stain and immunochemical positivity for S-100 protein and CD1a and negativity for macrophage markers.
Five cases of adult cutaneous LCH were noted in Korean Dermatologic literature (Table 1)4-8. Although LCH is commonly thought to be confined to children, a small percentage of patients are elderly and may have cutaneous involvement for many years before the onset of visceral disease9. Adult LCH has difference with childhood LCH in organ involvement. Bone disease is most common in childhood LCH, but pulmonary disease is very frequent in adult LCH. Skin disease and diabetes insipidus are frequent in both type10.
Cutaneous manifestations are common in multi-organ involved LCH and adult LCH skin lesions are similar that of childhood. The typical lesion is yellowish or reddish small multiple papules with ulceration, crusting or a hemorrhagic change on the trunk and scalp. Especially the scalp lesions resemble seborrheic dermatitis and other common finding is an eczematous lesion on intertriginous area11.
On the other hand, unusual cutaneous manifestations of LCH such as varicelliform eruption, solitary nodule and nail fold swelling are also reported (Table 2)12-19. In this case, cutaneous lesions are shown as brown lichenoid patches on left dorsal foot and we consider this is rare in LCH patients.
Surgery, steroid cream, topical nitrogen mustard, interferon IL, PUVA, isotretinoin, thalidomide can be treatment for skin manifestations of LCH. Single system-patients should receive prednisone and vinblastine for 6 wk followed by continuation treatment with 6-mercaptopurine, prednisone, and vinblastine for 6 months. Patients with multisystem disease should receive prednisone and vinblastine for 6 wk followed by continuation treatment with 6-mercaptopurine, prednisone, and vinblastine for 6 or 12 months, respectively20.
LCH presents a very wide clinical spectrum and variable course. Prognostic factors are including age of patients at diagnosis, numbers of organ involved and organ dysfunction. Single system disease is usually associated with a good prognosis, but multisystem disease may be fatal11.


 XML Download
XML Download