

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu disease, is an autosomal dominant disorder of the fibrovascular tissue1. It is characterized by the classic triad of mucocutaneous telangiectasias, arteriovenous malformations with recurrent epistaxis and hemorrhages, and inheritance2. Clinically it appears as punctuate or splinter-like telangiectasias located on the lips, oral mucosa, upper extremities, nail beds, and trunk3. HHT has rarely been reported in the dermatologic literature. We report a typical case of HHT in a 73-year-old female who had recurrent epistaxis and several skin and visceral manifestations.
CASE REPORT
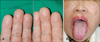
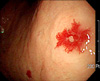
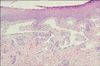
A 73-year-old woman was referred for evaluation of purpuric, punctuate, and tiny macules on the finger tips of both hands and the tongue which had been present for 50 years (Fig. 1). She had suffered numerous episodes of mild-to-severe nasal bleeding of unknown cause. She had been treated for epistaxis with electrocauterization therapy several times. She had a history of admissions to the Department of Gastroenterology due to exacerbations of anemia and melena 2 years before. During her cutaneous examination, we noticed pallor and discoloration with telangiectasias in the oral mucosa and tongue. The family history was significant for recurrent epistaxis and telangiectatic lesions in her mother and two sisters. No abnormalities were detected on chest x-ray. The laboratory work-up at admission revealed the following: WBC, 2,500/mm3; platelet count, 207,000/mm3; hemoglobin, 6.5 g/dl; ferritin, 3.72 (13~150 ng/ml); and serum iron 34 (50~150 ug/dl). Other laboratory work-up was normal, including bleeding time, coagulation time, prothrombin time (PT), activated partial thromboplastin time (aPTT), and stool occult blood. Endoscopy of the upper digestive tract was performed. The results of the endoscopy indicated multiple gastric angiodysplasias of the fundus and body of the stomach (Fig. 2). To prevent continuous bleeding and correction of chronic anemia, hemoclipping of multiple vascular lesions of the GI tract was performed as palliative treatment. We performed a punch biopsy from one of the macules on her left finger tip. The histopathologic study showed dilated capillaries lined by flat endothelial cells in the papillary dermis (Fig. 3). From these findings, the diagnosis of hereditary hemorrhagic telangiectasia was made.
DISCUSSION
HHT is a hereditary disorder with autosomal dominant transmission, despite the fact that about 20% of the cases do not have a family history. The reported incidence of HHT is approximately 1 per 5,000~10,000 population per year1. It is thought that the abnormal vessels in HHT develop because of aberrant TGF signaling at some stage during vascular development and homeostasis due to mutations of HHT-associated genes. There are two major types of HHT (HHT1 and HHT2). It has been proposed that in the case of HHT, disease severity is more pronounced in HHT1 compared to HHT2, with an earlier age of onset for epistaxis, the appearance of telangiectasias, and a higher incidence of pulmonary AVMs4. HHT1 can be induced by mutations in the gene, ENG (endoglin), encoding endoglin on chromosome 9q33,34. HHT2 can be induced by mutations in the gene, ALK-1 (activin receptor-like kinase 1), encoding activin receptor-like kinase 1 on chromosome 12q135. These events cause alteration in the elastic and muscle layers of vessel walls, making them more vulnerable to spontaneous rupture and injuries6.
The diagnosis of HHT is established when three of the following features are present: (1) epistaxis (spontaneous, recurrent nose bleeds); (2) multiple telangiectasias at characteristic sites (lips, oral cavity, fingers, and nose); (3) visceral lesions, such as gastrointestinal telangiectasia (with or without bleeding), pulmonary arteriovascular malformation (AVM), hepatic AVM, cerebral AVM, spinal AVM; and (4) family history (a first-degree relative with HHT)7. Our case met four criteria for HHT: recurrent epistaxis, telangiectasias of the fingertips and tongue, GI bleeding combined with anemia, and first-degree familial tendency.
The clinical manifestations of HHT are known to be variable and age-dependent1. Epistaxis is the first manifestation and the most common symptom, but patients may have a variety of serious complications due to vascular involvement of internal organs, such as the gastrointestinal tract, the lungs, and the central nervous system. In the follow-up of affected persons, the lung and brain are of particular concern, because each may contain clinically silent lesions that can result in sudden morbidity or death. Pulmonary AVMs, such as right-to-left shunts can result in hypoxemia. Furthermore, the absence of a filtering capillary bed allows emboli to reach the systemic circulation, which may cause cerebral abscesses and stroke. Cerebral AVMs can lead to headaches, migraines, brain abcesses, seizures, paraparesis, ischemia, strokes, transient ischemic attacks, and both intracerebral and subarachnoid hemorrhage. Gastrointestinal bleeding can result in iron deficiency anemia or acute gastrointestinal hemorrhage1. Vascular lesions may be present as telangiectasias, arteriovenous malformations (AVM), or aneurysms.
Treatment options for HHT should be considered individually for each patient, owing to the diverse clinical manifestations of this disease1. Therapy for the bleeding is primarily supportive and palliative. Telangiectasias of the skin and the mucosa are not merely a cosmetic problem, but may cause hemorrhages in >27% of patients, mostly involving hemorrhage from the tongue, the fingers, and the skin of the supraclavicular fossa, which worsen with age and alter the quality of life. Treatment should be offered to these patients. However, the awareness of the therapeutic necessities and specific methods of treatment were not adequate until now. Management options for cutaneous lesions include electrocauterization with diathermy, sclerotherapy, or laser therapy. Many different types of laser therapy have been used to minimize and/or eliminate these telangiectasias, such as Nd:YAG, IPL, argon, and the tunable dye lasers, all of which have been reported to be effective8.
Presymptomatic intervention in HHT may substantially affect the outcome. An early diagnosis is essential in high-risk individuals in order to alter their clinical course and prognosis. Patients with a family history of HHT with pulmonary disease are at high risk of having pulmonary AVMs. Chest CT-scan must be used and pulmonary angiography should be used for radiologic or surgical treatment planning. MRI is useful for assessing CNS involvement7.
Skin manifestations could be regarded as a minor condition of HHT, but the capacity for stigmatization must not be underestimated. It is important to recognize the skin manifestation of HHT for the early diagnosis of HHT to prevent complications. Moreover dermatologists should consider clinically similar telangiectactic diseases, including CREST syndrome (calcinosis cutis, Raynaud's phenomenon, esophageal dysfunction, sclerodactyly, and telangiectasia)7, hereditary benign telangiectases, and ataxia-telangiectasia as the differential diagnosis for HHT. Greater clinical awareness for dermatologists may be the first step to identify patients with HHT, and as such, may play a key role in the timely referral of these patients and their family members.
 XML Download
XML Download