

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Porokeratosis is a group of uncommon disorders of keratinization, either hereditary or acquired, that are characterized histologically by the presence of a column of paraketatotic cells known as the cornoid lamella1. Six clinical variants are recognized1. Among these six, disseminated superficial porokeratosis (DSP) is characterized by multiple small keratotic papules with central dell. The lesions may be erythematous or pigmented. Porokeratosis lesions are believed to result from the peripheral expansion of an abnormal, mutant clone of epidermal keratinocytes, an expansion that may be triggered by UV light, trauma, infection, or immunosuppressants1.
Inflammatory DSP is an unusual variant which was first described in 1992 by Kanzaki et al2, who called it eruptive pruritic papular porokeratosis (EPPP). The typical clinical course for patients with inflammatory DSP consists of several years of asymptomatic DSP followed by the appearance of intensively pruritic erythematous papules or plaques, which then subside within several months. Histologically the pruritic lesions display an infiltration of eosinophils and lymphocytes in the perivascular area of the upper dermis. Here we report an unusual case of inflammatory DSP that developed in a patient with colon cancer.
CASE REPORT
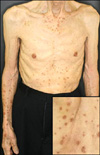
A 84-year-old man presented with a 3-month history of rapidly spreading pruritic eruptions over his trunk and extremities (Fig. 1). The patient had experienced asymptomatic, slightly elevated, brown-colored annular lesions on his trunk and limbs for 3 years. Suddenly, 3 months prior to presentation, pruritic lesions had developed on his arms and part of his trunk. He had a history of colon cancer, which had developed 6 years earlier and, which had been treated by a lower anterior resection of the descending colon and three cycles of chemotherapy. He had scattered multiple pigmented, hyperkeratotic annular lesions up to 3 cm in diameter on his arms and trunk,. There was no family history of DSP. Laboratory tests revealed eosinophilia (9.7%; normal range 0~5%); otherwise, all measured values were in the normal range.
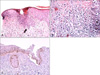
Histopathology performed on a skin biopsy from the central area of the lesion showed patchy parakeratosis, cornoid lamella, an absence of the granular layer below the cornoid lamella, and specific dermal inflammatory infiltrate with eosinophils and lymphocytes (Fig. 2A, B). A p53 immunohistochemical staining showed strong nuclear reactivity, mainly on the basal layer (Fig. 2C).
The patient was treated with oral antihistamine and a topical steroid for the management of pruritus, after which his pruritus subsided. One month after treatment there was no spreading of the lesions, and he was regularly checked for colon cancer.
DISCUSSION
Porokeratosis is an epidermal disorder with an autosomal dominant mode of inheritance and partial penetrance. Six clinical variants are recognized: (1) porokeratosis of Mibelli, (2) disseminated superficial porokeratosis, (3) disseminated superficial actinic porokeratosis, (4) linear porokeratosis, (5) porokeratosis palmaris et plantaris disseminate, and (6) punctate porokeratosis1. In 1992 Kanzaki et al2 reported three patients who developed an acute exacerbation of severe pruritus, and he named this atypical DSP "eruptive pruritic papular porokeratosis." In 1995 Tanaka et al3 reported another unusual DSP case, which they called "inflammatory DSP." The disorders referred to by these two terms follow the same clinical course and display the same histopathological findings4. According to our review of the published work, there have been eight cases of eruptive pruritic papular porokeratosis and inflammatory DSP reported in the literature including the case reported here5.
Inflammatory DSP patients generally exhibit a clinical course characterized by three stages. In the beginning they have asymptomatic DSP which lasts for several years; this is followed by the sudden appearance of intensively pruritic porokeratosis; and finally, after several months, these lesions spontaneously subside, leaving small brown spots or annular lesions. Our patient had asymptomatic DSP for 3 years and then pruritic symptoms for 3 months before presenting. But his pruritic symptoms remained despite the use of antihistamine medication and topical steroid for 3 months. All but one of the previously reported cases showed tissue eosinophilia without eosinophilia in the peripheral blood5. Our patient exhibited both tissue and peripheral eosinophilia.
Histopathologic findings for patients with inflammatory DSP show the stratum corneum to be hyperkeratotic, and a thin column of parakeratotic cells called the cornoid lamella is seen. The underlying keratinocytes are edematous with spongiosis and shrunken nuclei, and a dermal lymphocytic pattern may be evident. The granular layer underlying the cornoid lamella is either absent or reduced, but the granular layer in other areas has a normal thickness. The epidermis in the central portion of an area affected by porokeratosis may be normal, hypertrophic, or atrophic1, but inflammatory DSP shows additional eosinophilic infiltration in the upper dermis3.
Regarding the pathogenesis of porokeratosis, Reed and Leone suggested that a localized anomalous keratinocytic clone may lead to the epidermal dysplasia seen in porokeratosis6. The genetic basis for this porokeratosis is unknown, but several genetic loci have been identified to date. Among these loci, a genetic locus for DSP has been found at 18p11.37. Also, overexpression of p53 has been reported in porokeratosis8. Recently, frequent p53 overexpression has been reported and might be related to the carcinogenic potential of porokeratosis. In 2000 Takata et al9 reported a hereditary non-polyposis colorectal cancer associated with DSP. There are also a few reports of the development of DSP associated with two other cancers, hepatocellular carcinoma and cholangiocarcinoma (Table 1)10,11. Both reports suggest that an increased expression of the p53 tumor suppressor gene may have a crucial role for cancer and porokeratosis. Our patient had a history of colon cancer, but it is not clear whether an altered p53 system in the lesions might explain the codevelopment in the patient of colon cancer and DSP. Although we could not determine whether the onsets of colon cancer and DSP were related, immunologic aberrations and cytotoxicity may affect the biology of keratinocytes.
The treatment of inflammatory DSP is identical to the treatment of classic DSP. Intervention is usually unnecessary. However, if the lesions cause other problems or cosmetically unacceptable, treatment with a topical steroid, topical 5-FU, imiquimod, cryotherapy, a carbon dioxide laser, of a frequency-doubled Q-switched Nd:YAG laser can be considered12-15. Generally, pruritic symptoms will subside spontaneously within several months, but oral antihistamine can be useful if the patient has pruritus. There have been no reports of a malignancy developing from inflammatory DSP.
Since the first report, a total of eight patients, including ours, have been reported with inflammatory DSP or EPPP. We think that these two conditions are the same. Until now, there has been no report of inflammatory DSP being associated with a malignancy such as colon cancer. In this case study we have reported just such a rare occurrence, the case of unusual DSP in a 84-year-old man with a previous history of colon cancer.

 XML Download
XML Download