

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Clear cell sarcoma is a rare malignant soft tissue neoplasm that usually arises adjacent to tendons or aponeuroses1. It generally affects young adults with a predominance in women and mostly appears in the extremities, especially the feet and ankles2. The clinical course is rather slow, with repeated local recurrences followed by late metastases and eventual death3. Involvement of the abdomen is rare. We report a case of clear cell sarcoma affecting the abdomen.
CASE REPORT
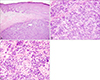
A 57-year-old female visited our clinic with a chief complain of a skin lesion on her right lower abdomen of two years duration. She had a mitral valve replacement surgery 5 years ago, and is now taking warfarin for maintenance therapy. Other than this, there is nothing special in her past medical or family history. In the physical examination we found a pea-sized glistening erythematous nodule on her right lower abdomen (Fig. 1). This nodule gradually increased in size but there was no pain or tenderness. We performed a punch biopsy on the tumor. The histologic examination revealed that the neoplastic cells were divided into well-defined nests and groups by fibrous tissue septa (Fig. 2). The cells consisted of round to ovoid vesicular nuclei and pale-staining cytoplasm. In the immunohistochemical staining the tumor cells showed positive reaction to S-100, Vimentin (Fig. 3) and negative reaction to Ki-67, CD34, SMA, Desmin. We performed a whole body PET-CT to ascertain the infiltrated depth of the tumor and the evidence of lymph node or distant metastasis. The PET-CT showed no other abnormal uptake except the 1 cm sized tumor in the right lower anterior abdomen. A radical excision was carried out and the resection margins were free of the tumor. There's no sign of recurrence for 14 months and now we are observing the progress of the disease.
DISCUSSION
Clear cell sarcoma (CCS), first described by Enzinger1 in 1965, is a rare soft tissue tumor. Before this, this uncommon neoplasm had been misdiagnosed as fibrosarcoma, synovial sarcoma, hemangiopericytoma, alveolar soft-part sarcoma, and hemangioendothelioma4. It occurs most frequently in the feet and ankles of women in the second and third decades1. The tumor is characterized by multiple local recurrences with late metastases and a high rate of deaths3. It usually presents as a slowly growing mass and occasionally causes mild pain or tenderness2. Symptoms may persist for a long time (mean of 5 years) before the patient seeks medical attention2.
Gross pathologic examination of CCS reveals a localized, tan-gray, firm and somewhat circumscribed mass, varing in size from 0.4 cm up to 14.5 cm5. Frequently, the mass is attached to tendons or aponeuroses, but there is no direct connection with overlying skin6. On microscopic analysis, epithelioid cells with pale nuclei and deeply basophilic nucleoli are aggregated in compact nests surrounded and separated from adjacent nests by fibrous tissue septa7. Mitoses are generally sparse and necrosis and haemorrhage are rare8.
On immunohistochemical examination, the tumor cells of nearly all cases express S-100 protein3. Most of them also express antigens associated with melanin synthesis (HMB-45, melanin-A, Mel-CAM)3. Cytokeratin, epithelial membrane antigen, carcinoembryonic antigen, desmin, and smooth muscle actin are usually negative9. In our case, staining the tumor cells showed positive reaction to S-100, Vimentin and negative reaction to Ki-67, CD34, SMA, Desmin.
Ultrastructural analysis shows tumor cells closely apposed to each other by continuous basal laminae and rudimentary cell junctions9. Cells show abundant cytoplasm containing numerous mitochondria and aggregates of glycogen9.
The differential diagnosis includes synovial sarcoma, fibrosarcoma, epithelioid forms of malignant peripheral nerve sheath tumor, spindle cell melanoma. One of the main differential diagnoses of CCS is malignant melanoma (MM). CCS and MM demonstrate significant morphologic overlap at light microscopic and ultrastructural levels, as well as similar immunohistochemical features, so the distinction may be difficult5. However, the behavior of CCS is decidedly different from that of MM, demonstrating a more indolent course7. CCS is a deep seated tumor that rarely shows the degree of anaplasia, necrosis, and mitotic activity usually associated with MM. Morever the t(12 ; 22) translocation and EWSR1/ATF1 gene rearrangement observed in the majority of cases of CCS have never been found in MM5. Approximately 50% of MMs showed deletions of short arm of chromosome 9 at the interferon alfa locus5.
Treatment should be radical resection of the tumor, followed by chemotherapy and radiotherapy2. However chemotherapy and radiotherapy have not been shown to be of benefit510, so early recognition of this disease and prompt wide excision of tumor is essential for a favorable outcome11. The prognosis for patients with CCS is generally poor, although those with tumors smaller than 2 cm have a better prognosis12. Poor prognosis is associated with tumor size more than 5 cm, presence of necrosis, metastasis and local recurrence5.
We encountered a rare case of CCS arising in the abdomen. Although CCS is usually fatal after a long clinical course, our patient is still alive without recurrence 14 months after resection.


 XML Download
XML Download