

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Incontinentia pigmenti (IP), also known as Bloch- Sulzberger's disease, is a rare X-linked dominant genodermatosis caused by a mutation in nuclear factor-κB (NF-κB) essential modulator (NEMO) gene on the X chromosome, localized to Xq28. IP predominantly affects female infants (in excess of 37:1), and is usually lethal in males in utero12. The survival of affected males is attributed to the presence of an extra X chromosome (Klinefelter's syndrome), hypomorphic mutations, and somatic mosaicism345. Only two cases of IP in male infants without karyotype study have been reported in the Korean dermatologic literature since 198267. This study reports a rare case of incontinentia pigmenti in normal karyotype (46, XY) male infant.
CASE REPORT
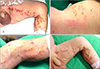
A 3-day-old boy was referred to our department for the evaluation of skin finding presented at birth. The lesion showed erythema, linear vesiculations, and focal crust on the trunk and leg, predominantly over the left side (Fig. 1A, B).
At the time of birth, several vesicles and erythematous papules developed over the left trunk and thigh along Blaschko's lines. Blistered skin was replaced by a crusted lesion on the fifth day after birth (Fig. 1C). Then, hyperkeratotic papules and verrucous lesions occurred consecutively on the fifteenth day (Fig. 1D).
The patient was the second child born on normal full term delivery to the patients. His parents and his elder brother were healthy, and had no other comparable dermatoses.
There was no remarkable abnormality except for the skin lesions on physical examination. Laboratory tests including complete blood count, blood chemistry analysis, urinalysis, VDRL, bacterial culture, and TORCH serology (for toxoplasmosis, rubella, cytomegalovirus and herpes simplex) were in the normal range or negative. Also, radiologic findings in brain MRI, echocardiogram, and chest, abdomen, both extremities X-rays appeared normal. Chromosome analysis from peripheral blood leukocytes showed a normal male karyotype (Fig. 2), nonetheless, due to the refusal of parental consent, molecular analysis was not executed.
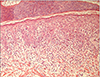
A skin biopsy was taken from one of the blistered areas on the left thigh. Histopathologically, epidermal hyperkeratosis, spongiosis, dyskeratotic cells throughout the epidermis, basal liquefaction degeneration, and inflammatory infiltration composed predominantly of eosinophils were noted (Fig. 3).
The patient was diagnosed as IP, and received antibiotic treatments to control secondary infection. After 2 months, the lesion disappeared with residual hyperpigmented streaks on the left thigh.
DISCUSSION
Although a case resembling IP was first described by Garrod in 1906, IP was characterized as a clinical syndrome by Bloch in 1926. In 1927, Sulzberger reported Bloch's case in details, and then proposed as Bloch-Sulzberger's disease2.
In general, boys with single X chromosomal gene abnormality are so severely affected as to be lethal in utero, therefore there is an increased incidence of spontaneous abortion in affected women, presumably of male fetuses89.
The major cause of IP is loss-of-function mutation of NEMO protein, most consisting of a deletion of exons 4~10 in heterozygous females310. The gene mutated in IP is mapped to Xq28 and encodes the NEMO. NEMO is a regulatory component of the IKB kinase complex required for NF-κB activation. Therefore, it is central to many immune, inflammatory responses, and apoptotic pathways. Disruption of NEMO gene leads to diminished NF-κB activity which increases the susceptibility of cells to apoptosis1112
It is quite rare for male patients with IP to survive. Survival occurs in approximately 2~3% of all the reported cases13. It can be explained by three potential mechanisms: abnormal karyotype (Klinefelter's syndrome, 47, XXY), hypomorphic mutations, and somatic mosaicism. The third is the most reliable explanation for the survival of male patients. It results from a postzygotic mutation occurring during the blastocyst stage of embryogenesis and does not completely abolish NF-κB activity, which allows survival34514. Thus, the presence of somatic mosaicism results in mild features of known genetic disorders, and better outcomes afterward15. Skin findings of live born male patients are generally not greater than those in affected females and many male patients have disease expression limited to cutaneous involvement of one or two limbs2. According to Scheuerle9, live born males with IP are inferred to be of generally good health and not at increased risk of neonatal or infantile mortality, because multiple hospitalizations or life threatening illness have not been reported. The clinical features of IP are associated with ectodermal tissue abnormalities including skin, hair, nails, teeth, eyes, and central nervous system. Skin finding is characterized by typical linear lesions subdivided into 4 stages with variable timings: Perinatal inflammatory vesicles (stage 1), verrucous patches with hyperkeratosis (stage 2), a distinctive pattern of streaky hyperpigmentation following Blaschko's lines (stage 3), and finally atrophy, hairlessness, and scarring (stage 4)1316. These represent the death of cells carrying the mutated gene along lines of embryonic cellular migration12. However, some of the stages may occur concurrently with others, or not at all.
In addition, females who carry loss-of-function mutations would be expected to often have severe clinical signs, including neurologic: spastic paralysis, convulsion, and mental retardation; ophthalmologic: strabismus, cataracts, microphthalmos, cataract and optic atrophy; and odontologic defects: partial anodontia, delayed impaction, and crown malformations, presenting at birth or during the first few weeks of life10111314.
The severity of IP is related to ocular and neurological impairment, in particular, blindness and psychomotor retardation, not skin lesions117. The latter generally disappear spontaneously, and no treatment is necessary other than the control of secondary infection. However, other complications should be sought carefully and, if possible, treated quickly as soon as the diagnosis of IP is established. Also, urgent consultations with ophthalmic, neurologic and dental subspecialists are often needed.
In conclusion, in a survival case of male infant with IP, careful evaluation, chromosomal analysis, and family counseling should be performed to rule out alternative diagnoses and for better clinical outcomes.

 XML Download
XML Download