

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Stem Cells (SCs) biology has given a way toward the utility of regenerative medicine and methods in the isolation of tissue-specific SCs. Tissue-specific SCs are important for retaining tissue homeostasis and wound healing processes. These SCs have two basic functional characteristics: self-renewal and differentiation potentials in cell lineages. In early SC technology to assess these characteristics, they found out cell differentiations were not equivalent and some cells seemed to have more activity in proliferation at different differentiation stages. By further observations, they hypothesized that the existing proliferating undifferentiated cells might differentiate into self-renewal potent cells [1].
By virtue of the development in functional assays including in vitro clonogenic assays and lineage-tracing experiments, the differentiation of multilineage in tissue-specific SCs was suggested. This suggestion was primarily demonstrated by bone marrow cells transplanted in irradiated mice, which differentiated into different haematopoietic lineages. Later, there was an experiment using epithelial cells that showed that keratinocytes stimulate the differentiation. These cells were engrafted into long-term renewal cells as functional tissue [2]. This is meaningful in the in vitro cultivation of SCs for maintaining differentiation potential. In a more advanced research, in vivo lineage tracing has been adapted to demonstrate the crucial role of SCs during tissue homeostasis and repair in their natural environment [3].
SCs have self-renewal ability, which is the most useful hematopoietic property of tissue regenerative applications. Perhaps the origin of cancer is from the transformation of normal SCs, cancer cells in a given tissue also do not have identical properties with each other like in SCs. There exists a small portion of cancer cells, which have rapid differentiation potential compared with others. Not like SCs, these cancer cells have few unique characteristics. They have potency to generate tumor, in vivo propagation, and potential to generate different self-renewly potent cells with variant phenotypes [4]. Taking the similar signaling pathways of the ability to self-renew in cancer cells, several rare cells with indefinite potential that drive tumorigenesis, are known as cancer stem cells (CSCs). Their origin is yet to be discovered, though a series of hypotheses have been proposed in this regard.
The concept of CSCs derives from the fact that cancers are dys-regulated tissue clones that continuously maintain propagation from a biologically distinct subpopulation of cells which are very uncommon [5]. This concept was recently recognized as a widely accepted rationale because of the expanded definition of hierarchies in normal tissue, multistep traits of oncogenesis, and improv-ed methods of xenotransplantation that from primary human cancers in the immunodeficient mice model.
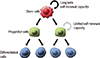
There are two concepts to define: heterogeneity and cancer cell proliferation. The first concept explains that the cancer cells are equivalent and a proportion of cancer cells proliferate and differentiate stochastically. Compared to the first concept, the second concept explains that cancer cells are hierarchically organized, that certain cells are involved in cancer growth and that undifferentiated progenitor cells have limited growth potential (Fig. 1). The similar model of tissue-specific SCs supports the second concept of CSCs that are responsible for the tumor growth and maintenance of cancer. Both these concepts are further extended to clonal evolution, in which cancer cells progressively accumulate genetic alterations such as base substitutions and small insertions/deletions. Some of which provide increased advantages of fitness and survival and allow the mutated cancer cells to outcompete other cancer cells [6].
In a recent study, evidence has shown that some cancer cells shar-ed certain molecular mechanisms like in tissue-specific SCs [7]. An increased understanding of the molecular signaling events that regulate cellular hierarchy and stemness, and successes in defining molecular signatures of CSCs have opened up new avenues that have accelerated the development of novel diagnostic and treatment strategies. Here, we highlight the recent advances in understanding of the pathogenesis in CSCs by comparing different techniques of functional assays to reveal genetic heterogeneity and proliferation hierarchy. Furthermore, we also discuss the implications for targeted therapy in cancer cells.
HETEROGENEITY IN NORMAL TISSUE AND CANCER
Heterogeneous potential was initially introduced by the demonstration of epithelial tissues in the epidermis. Through the different culturing conditions on keratinocytes, the three types of clones have been identified and they had shown different clonogenic potentials of the epidermis. The holoclones consisted of undifferentiated cells including epidermal SCs and showed the highest proliferative capacity. On the other hand, the meroclones were in the transitional stage, and the paraclones had transit-amplifying progenitors that showed terminal differentiation [8]. Moreover, they showed holoclones presenting considerable proliferative potential in vitro, but most of these cells were quiescent in vivo [9]. Finally, the idea that these cells are multipotent SCs were derived from transplantation and isolation using microdissection and flow cytometry [10].
To look into the tissue heterogeneity, in vitro clonogenic assays were applied to study differentiation potentials of different cell populations at a single-cell level. The sphere-forming assays were adapted in SCs and CSCs to assess the multilineage differentiation and renewal capacities. But, they were restricted only in a fraction of cells from primary colon [11], breast carcinoma [12], and brain carcinoma [13]. Since only these tissues were able to form spheres, accurate quantification of the differentiation frequency is not grant-ed in SCs in the in vivo system. Recently, to expand SCs and progenitor cells from different epithelium, the organoid culture method (three-dimensional, non-adherent condition) was applied [14].
The same strategies were applied on CSCs to explain the different growth potentials that contributed different cancer growth in the microenvironment such as xenotransplantation and in vitro [15]. The experiment in mouse glioma had shown that the group of cells which had low expression level of inhibitor of DNA binding 1 (ID1) generated secondary cancer more efficiently than the group of cell which had higher expression level of ID1 on transplantation, but the group of ID1low cells presented lower self-renewal potential than ID1hi cells in a sphere-forming assay in vitro [16].
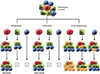
Serial transplantations of cancer cells on sphere form in vitro assess the self-renewal potential of CSCs. A large amount of isolated cells were required to maintain secondary cancer cell population, because most studies precluded the assessment of their self-renewal ability after transplantation. The gold-standard assay to this problem was solved by the genetic lineage-tracing approach (Fig. 2). It explains self-renewal and differentiation capacity of cancer initiation and cell development. In mouse genetic lineage-tracing experiments, transgenic mice expressed a reporter gene by drug-inducible Cre activation in a specific lineage. This experiment came out with different types of differentiated cells referred to the differentiation potential of SCs that the fraction of indicated cells persisted over time [17]. In mouse squamous cell carcinoma, the frequency of tumor propagating cells (TPCs) was increased in CD34hi population but decreased in CD34low population during serial transplantation, whereas there was no change in primary cancer [18]. This was also shown in mouse lung adenocarcinoma, the frequency of TPCs was enriched only in the spinocerebellar ataxia type 1+ cells. However, the spinocerebellar ataxia type 1- cells only gave rise to small secondary cancers that could not be serially grafted [19].
Although the development of the sphere-forming assays had a great role in finding the characteristics of CSCs in vitro on single cell levels, there are still unrevealed questions about what extent of the capacity of cancer cells growing as spheres directly correlated with sustained cancer growth in vivo.
BIOLOGICAL AND MOLECULAR CHARACTERISTICS OF CSCs
Since normal SC has no universal molecular characteristics, we can use biological markers like LGR5 to identify different populations of SC in the specific tissues. The same aspects was applied to CSCs in that CD133 had been associated with TPCs in many different types of cancer tissues [20]. Using experimentally confirmed surface markers, CSCs' population can be assessed successfully. In liver cancer, cell markers like CD133, CD90, CD44, CD24, CD13, and epithelial cell adhesion molecule were used to identify the liver CSC, which had features of self-renewal potential and tumor growth [21].
Based on the signaling pathway of regulating normal SCs, when the WNT-β-catenin signaling pathway was abnormally activated with nuclear factor-κB, it was influenced in intestinal epithelium shown by the occurrence of colorectal cancers [22]. This is consistent with the idea that the signaling pathway of normal SCs can induce cancer initiation by reprogramming to a progenitor-like fate [23]. This suggests that the role of CSCs have similar features with tissue development on the activation of signaling networks that leads to cancer development.
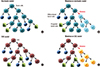
Clarifying the mechanism of cancer initiation in various types of cancer is necessary to augment molecular features of CSCs. Through experiments on transplanting CSCs into immunodeficient mice, these cells are defined to reform secondary cancer operationally. In colorectal cancers, when the WNT-β-catenin signaling pathway was mutated on transplantation, active β-catenin was over-regulated and it gave CSCs properties to LGR5+ cells by dedifferentiation on secondary cancer [24]. Similarly, in skin squamous cell carcinoma, CD34low cells gained CSCs potentials from the transplantation of CD34hi cells. However, the differentiation by CD34low cells in secondary cancer does not directly imply the efficiency of CD34hi cells isolated from the primary cancer [18]. In human breast cancers, the non-TPCs presented a great population of CD44hi CD24-marked cells in secondary caner, which means they have gained CSCs potentials. These results suggest that a transition of stochastic state is associated with increased the potential of CSCs among different subpopulations [25]. To verify this suggestion, more research based on the transition of stochastic state occurring in primary tumors from different tissues will be needed (Fig. 3).
Not only cancer initiation, but cancer progression and metastasis are also important features in CSCs. Usually the metastasis by CSCs was achieved through producing mutants by the stochastic state transition-associated genes. For example, in colorectal cancer, the CD26+ cells induced liver metastasis more than CD26- cells, and long-term TPCs were able to perform metastases [26]. These cells have been known as a subset of CSCs. Other evidence of metastasis were also found in prostate cancer [27], breast cancer [28], and melanoma [29] by expression of CXCR4, which was expressed in pancreatic adenocarcinoma from CD133+ CSCs [30]. But this is still controversial as to whether CXCR4 is related to CSCs in other human metastatic cancers.
Therefore, in case that a study can determine the cellular hierarchy of cancer metastasis from CSCs, the cancer cells expressing certain CSCs markers like CXCR4 or CD26 could be potential therapeutic targets by pharmacological inhibition.
MICROENVIRONMENT AND CSC NICHE
The microenvironment can reflect the heterogeneity of SCs and CSCs that might utilize the balance between self-renewal and differentiation features. This specific native environment is called a niche; the secretion of soluble molecules or cell-to-cell communication balances the heterogeneity of SCs. To identify the actual progenitor-like potential of SC, it is important to recognize that SCs present an important plasticity in vivo. Under SC depletion, these progenitor-like cells present a certain degree of plasticity that reorganizes the SC niche and clonial repopulation [31].
For cancer initiation and cancer growth, it is obvious that angiogenesis and secretion of growth factors from endothelial cells are very important. For this reason, the regulation of perivascular niche can be the main key to regulate CSCs. This has been shown in normal SC that specialized the composition of mesenchyme in their niche, such as neuronal SC with endothelial cells that secreted growth factor to utilize the self-renewal features [32]. However, the components of cancer niche remain generally unknown. One experiment showed CSCs were related to endothelial cells in glioblastoma by culturing CSCs in vitro that greatly utilized their self-renewal potentials. Which claimed that perivascular niche enhanced the formation of CSCs in glioblastoma [33]. Another similar experiment can be found in malignant brain cancer. They found that therapy using anti-vascular endothelial growth factor A (VE GFA) had reduced the population of CSCs [34]. Accordingly, the density of CSCs was also decreased in squamous cell carcinoma by inhibiting angiogenesis with VEGF receptor 2 antibodies. It has been recently reported that VEGFA stimulates the formation of perivascular niche that indirectly enhances the formation of CSCs [35].
There was an opposite view on the perivascular niche. One has reported that the hypoxic niche increased the clonogeneity of SC. In hematopoietic normal SCs, the leukemic SC population has been decreased under the absent condition of hypoxia-inducible factor (HIF) 1α in vivo [36]. This means the hypoxic niche promotes HIF to form an environment that is allows SCs to manipulate stemness in cancer cells. Moving on to hypoxia in CSCs, HIF1α activates the expression of CD133+ cells in pancreatic cancer and estrogen receptor (ER)+ cells that expand TPCs in breast cancer [37]. These results show that different CSCs responded differently to hypoxia conditions. Although the regulation of CSCs in two different models, the perivascular niche and hypoxic niche seem to have no connections, but one niche can transform to another by abnormal functionality.
Another study on the regulation of CSCs has found a close association between fibroblasts and colorectal cancers, and that myofibroblasts activated in WNT signaling stimulate the cell to cell communication between CSCs [38]. In liver cancer, usually the capacity of proliferation of the hepatic SCs is increased in chronic liver disease. The hepatocyte proliferation is induced by continuous cell regeneration but the proliferative capacity, which is affecting motility and proliferation, is not infinitely increased. In the CSC niche, the activation of hepatic progenitors is tightly regulated and related to other risks of hepatocellular carcinoma (HCC) like liver infection and inflammation [39]. HBV and HCV infection promote cell mutagenesis and genomic instability resulting DNA damage to induce HCC [40]. The HBV X gene study suggested that the HBV X gene promoted HCC by modulating p53 signaling pathways [41]. Similarly, inflammation might give rise to oncogenic events resulting in cell proliferations and genetic alterations. [42]. Therefore, accumulating the pathophysiological changes in a range of liver lineages would verify the development mechanism of liver CSC.
Further understandings of these molecular interactions and the cellular factors of the niche are needed to identify the genetic and epigenetic mechanism of cancer clonal evolution. Defining the contribution of the establishment of CSCs would then allow extension into targeted therapy of CSCs on cancer propagation.
CLINICAL APPLICATIONS ON GENETIC TARGETED THERAPY
The main focus of targeted therapy on CSCs is the capability of cancer relapse. Just like normal SCs, they are also resistant to chemotherapy, radiotherapy, and other therapies. After radiotherapy in xenografts using TPCs of glioma, one showed that CSCs rapidly repaired damaged-DNA and avoided apoptosis induced by ionizing radiation [43]. Another study also demonstrated in the colorectum CSCs initiated the activation of damaged-DNA checkpoints [44]. Also, radiotherapy in brain cancer was affective when blocking the activation of checkpoint kinases CHK1 and CHK2 from CSCs [43]. In breast cancer, because of high expression of the free radical-scavenging mechanism in SCs, the inhibition of scavenging machinery decreased its clonogenicity and could give affective radiotherapy [45].
In clinical applications, there are two ways to improve cancer treatment. One is the development of novel cancer diagnosis, and the other one is development of new strategies of targeted treatment. For example, gene expression profiling techniques by using microarray or next generation sequencing can help identify dys-regulated genes in cancers that reveal tumorigenic, metastatic features, and/or predict patients' prognosis. As we already know, early diagnosis of cancer is very crucial and will have great effect on patient survival. However, conventional diagnosis on the cancer tissue has limitations on predicting the actual prognosis change after radical treatment. By applying gene expression profiling technologies, these limitations were resolved in liver cancer [46]. In the future, CSCs could be isolated very easily from circulating tumor cells in peripheral blood, and may provide diagnostic or prognostic impacts.
Generally, HCC patients are treated with transcatheter arterial chemoembolization (TACE) in the intermediated stage. But there is the problem of tumor cells relapsing from CD133+ CSCs after TACE. To prevent HCC recurrence, 5-FU or CD13 inhibitor are applied for targeting CD133+ CSCs [47]. By advanced examination, a study suggested that 5-FU inhibited the signaling pathway of PTEN, Akt, and ABCG2 to suppress the self-renewal ability of CD133+ liver CSCs [48]. The therapeutically beneficial model of targeted therapy to control cancer growth would directly eradicate CSCs. This therapeutic approach has been performed with patients in acute promyelocytic leukemia by all-trans retinoic acid controlling self-renewal and differentiation [49]. To enhance the targeted therapy of CSCs, it is important to identify the cellular factors that inhibit the growth of CSCs without affecting normal SCs. Recently, numerous methods to develop the library of these cellular level molecules have been found by screening techniques. Through classical chemotherapy in mouse glioblastoma, cancer-relapsing cells were identified and then the rate of cancer relapsing, targeting were decreased using genetic lineage ablation approach to these cells [50]. Therefore, more specific research is demanded on therapeutics focusing eradication of CSCs, and it may lead to a considerable clinical benefit.
CONCLUSION
The transplantation of CSCs to immunodeficient mice is mainly focused on applying the highest clonogenic potential that have no effect on any medical therapy. The genetic and epigenetic pathways of clonal evolution in metastasis are still not clear on whether the population of cancer cells are independently formed or evolved in branching. To provide an essential insight to this remaining uncertainty, the following question is necessary to be answered on other clonal analyses. Does in vivo lineage ablation technique maintain the potential strength of CSCs compared to transplantation assays? Many studies have shown significant results by lineage-tracing experiments in mice CSCs. To verify these results on human cancer, it is important to develop new techniques like viral barcoding and inducible lineage tracing approach. The first step is to build mouse models that provide the same niche found in primary human cancers.
In the field of targeted therapeutics, the advancement of novel assay would play a great role in identifying genetic heterogeneity and cancer hierarchy of CSCs. By finding the irreversible transformation between normal SC into CSCs, an appropriate targeted therapy will be developed to prevent cancer initiation and relapse after targeted therapy.
 XML Download
XML Download