

 PDF
PDF ePub
ePub Citation
Citation Print
Print



서론
비만 관련 질환은 서구사회에서 사망원인의 큰 축을 형성하고 있을 뿐만 아니라 미국에서는 성인의 65%가 과체중이며 약 1/3이 비만으로 집계되고 있다[1]. 우리나라에서도 비만은 서구사회만큼 중요한 문제로 인식되고 있으며, 매년 남자에서 2.8%, 여자에서 4.2%씩 증가하고 있다. 대사증후군은 단일 질환이라기보다 내장 비만, 지질 이상혈증, 고혈당, 고혈압 등 일련의 질환군으로 복합적으로 정의되는 궁극적 의의는 2형 당뇨병 및 심혈관 질환의 고위험군을 조기에 인지하는데 도움을 주기 때문이며 이를 적극적으로 치료해야 한다. 대사증후군의 기전으로 가장 중요한 것은 인슐린 저항성이고, 이외에도 염증 유발성 사이토카인, 지방세포에서 유리되는 adiponectin 등을 들 수 있다[2]. 그러나 아직도 단일 유발인자가 밝혀지지 않고 있어 그 원인이나 병태생리에는 논란의 여지가 많다. 특히 비만의 기전을 단순히 열량의 섭취와 소비의 불균형에서 찾는 시도는 유효한 치료적 전략을 제시하는 데에 어려움이 있기 때문에 타당하지 않은 것으로 여겨진다[3].
대사증후군은 비단 성인질환만이 아니라 소아에서도 중요한 질환군으로 인식되고 있다. 미국에서 소아비만은 20%에 달하고, 비만이 있는 소아의 85%가 성인비만으로 이어진다는 것은 그 중요성을 시사하는 바가 크다[4]. 1세 미만의 유아에서 성인기 비만으로 이어질 위험도(odd ratio)가 1.3정도이며 청소년기에서는 17.5까지 증가한다[5]. Weiss 등[6]의 연구에 의하면, 소아에서도 대사증후군이 흔히 진단될 수 있고, 인슐린 저항성 및 비만 등과 강하게 연관되어 있으며 C반응 단백이나 인터루킨(interleukin)-6와 같이 염증 유도 사이토카인이 관여한다고 밝혀져 대사증훈군이 소아기때부터 발생할 수 있음을 시사하였다. 흥미로운 것은1990년대 일련의 연구들은 대사증후군이 훨씬 더 일찍, 심지어 태아에서부터 기원할 수 있다고 보고하였다[7]. 따라서 본 글에서는 비만을 포함하여 대사증후군의 발달학적 측면에 대하여 살펴보고, 이를 통하여 신생아기부터의 대사증후군이 지니는 의의에 대해 알아보고자 한다.
산모의 영양과 태아의 절약 형질
산모의 영양상태는 태아에게 직접적인 영향을 미치게 되는데 산모가 비만할 때는 물론 영양이 부족할 때 모두 부정적으로 작용하게 된다. 산모가 비만하면, 임신 후반기에 특히 인슐린 저항성이 증가하게 되어 태아의 영양과다로 이어져 체중이 커지고, 체내 저장이 늘어나 출생 후 각종 이환율의 증가로 이어진다[8]. 동물연구에 의하면 고지방 식이를 한 어미쥐에게서 난 새끼쥐들이 더 비만하고 혈중 콜레스테롤이 높으며 혈압도 높아 성인기의 대사증후군 상태를 나타내었고, 유리 지방산의 농도 및 중성 지방의 농도도 높고 인슐린 저항성도 증가하는 것으로 보였다[9].
반면에 산모의 영양이 부족할 경우에도 태아에게 대사증후군의 위험도를 높일 수 있는데, 산모의 영양결핍으로 태아는 체중이나 신장이 작은 자궁 내 발육 지연(intrauterine growth restriction) 상태가 된다. 자궁 내 발육 지연 상태에서 태아는 열악한 영양 환경 조건 하에서 살아남기 위해 적응을 해가는 과정에서 성인기에까지 특징지어지는 어떠한 비전달성(noncommunicable) 형질을 발달시키게 되는데, 이러한 가설을 Barker 등[10]과 Hales와 Barker [11]는 '절약 형질(thrifty phenotype)'로 설명한다. 절약 형질 가설은 태아가 열악한 조건에 있을 때, 뇌나 심장의 중요한 기관을 보호하기 위해 장관 등의 덜 중요한 장기는 상대적으로 희생시킨다는 것인데, 특히 인슐린 저항성을 증가시킴으로써 뇌조직을 보호하게 된다는 것이다. 따라서 생존 가능성은 증가하지만 췌장의 베타 세포가 손상을 입어 당뇨병의 위험이 높아진다. 이에 따르면 2형 당뇨와 같은 만성질환들이 유전적 비중보다 오히려 태아기의 환경에 굉장히 민감하게 작용할 수 있을 것으로 생각된다[12]. 이에 대한 증거로서 Hales 등[13]은 1세 이하에서 체중이 적을수록 당불내성 및 인슐린 저항성이 증가한다고 보고하였고, Barker 등[10]은 64세의 노인들을 대상으로 시행한 후향적 연구에서 과거 출생체중이 2.95 kg 미만이었을 경우에 4.31 kg 이상이었을 경우보다 2형 당뇨가 10배 이상 많았다는 것을 확인하였다. 과거 출생체중이 작을수록 이환율이 높았던 것은 2형 당뇨뿐 아니라 인슐린 저항성, 고혈압 및 허혈성 심혈관 질환에서도 같은 경향이 관찰되었다[1415]. 출생체중과 혈중 포도당 수치 및 인슐린 대사의 연관성을 보기 위한 메타분석에 의하면 출생체중이 작을수록 금식 후 당내 불내성이 증가하고 포도당 부하검사에서도 혈당이 높았으며 2형 당뇨가 흔하고 인슐린 저항성이 증가하였다[16]. 이러한 형질의 특징이 비전달성이라는 연구를 뒷받침하는 최근의 연구로서 Han 등[17]은 비만의 대표적인 37가지의 단일 염기 다형 분석(single nucleotide polymorphism)을 비교 분석하였는데, 출생 시 체중이 작은 부당 경량아(small for gestational age)에서 소아비만이 발생할 때, 유전적인 요인보다는 비유전적 및 환경적 요인이 더 중요하다고 결론지었다.
절약 유전자형과 후성적 유전
지금까지 기술한 대사증후군의 발달학적 측면에 대한 고찰은 세대를 통해 유전되기 보다는 개체별로 획득되는 후성적 요인에 대한 것이었으나, 비만, 대사증후군이 세대를 통해 유전되는 가계가 존재하므로 이에 대한 고찰도 필요할 것이다. 앞서 설명한 Halse 및 Barker 등의 절약 형질 가설과는 달리, Neel [18]의 절약 유전자 가설(thrifty genotype)에 의하면 풍년과 기근이 반복되는 환경에서는 식량이 풍족할 때 열량의 저장을 늘려서 기근을 대비하는 선택적 생존으로서의 절약 유전자가 초래된다고 하였다. 이 유전자는 음식을 섭취하고 그 열량을 사용하는데 극도로 효율적이지만, 기근이 없이 풍년만 지속될 때에는 체내의 영양축적이 과해지므로 비만과 당뇨에 취약해질 수밖에 없는 것이다[1819]. 이에 대한 증거들로는, 과거에 수렵과 채집을 통해 주식을 해결하던 북미의 인디언 족에서는 좀처럼 비만을 찾아보기 어려웠으나, 영양적으로 풍족한 현대 사회에 이르러 비만과 당뇨의 유병률이 급격하게 증가하게 된 것을 들 수 있다. 이러한 특징은 이스라엘로 집단 이주한 에티오피아계 유대인에게서도 나타났고[20], 극심한 영양 결핍을 겪었던 2차 세계대전을 전후로 남태평양의 나우루 지역에서 보였던 당뇨병 유병률의 변화에서도 동일하게 나타났다[21].
개체별 후성적으로 획득한 대사증후군의 형질도 다음 세대로의 유전(epigenetic inheritance)이 가능하다. 이러한 양상을 라마키안 유전(Lamarckian inheritance)이라고 하는데, 멘델리안 유전(Mendelian inheritance)과는 구별된다[22]. 유전자 각인(genetic imprinting)은 DNA의 cytosine 기에 메틸화가 이루어지면서 형성되는데, Xist, H19, IGF2R 및 인슐린 등에 변형을 초래한다[23]. 이와 유사한 각인의 과정이 미토콘드리아 전사 인자 A의 유전자 부호화에 영향을 미쳐 미토콘드리아 DNA (mitochondrial DNA, mtDNA)의 수적 열세와도 연관이 되어있는데, Lee 등[24]에 의하면 27쌍의 산모 및 신생아를 대상으로 한 연구에서 말초 혈액에서 mtDNA의 수가 적은 산모일수록 신생아의 체중이 작은 것을 확인하였다. mtDNA에 변이 혹은 수적 열세로 인해서 기능에 문제가 생기면 지방산의 출입에 영향을 주어 β oxygenation에 장애를 초래한다. 그 결과 지방형성능 및 중성지방 합성을 증가시키게 될 뿐만 아니라 인슐린 저항성을 유발하여 당뇨를 초래하게 된다[25]. 따라서 이를 고려해 볼 때, 후성적으로 획득한 절약 형질은 모체를 통해 다음 세대로 유전될 수 있는 기전에 중요한 단서가 될 수 있으므로 이에 대한 후속 연구가 필요하다.
인슐린 저항성 획득의 기전
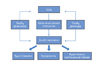
인슐린 저항성은 대사증후군의 병태 생리에 있어 가장 핵심적이다. 인슐린 분비를 담당하는 췌장의 베타 세포는 생후 1년 동안 성인 수준의 약 절반 정도까지 생성되기 때문에 베타 세포는 태아 및 신생아 시기가 매우 중대한 시기라고 할 수 있다[26]. 이러한 시기에 영양결핍이나 유전자적인 문제가 인슐린 분비 및 민감도에 문제가 생길 수 있다는 의미에서 태아 인슐린 가설을 들 수 있다[7]. Ozanne 등[27]은 저단백 식이를 한 어미 쥐에게서 획득한 새끼 쥐의 근육에서 glucose transporter 4에 의한 포도당의 이동에 중요한 역할을 하는 protein kinase C의 의미 있는 감소를 증명하여 태아에서부터 인슐린 저항성이 발생하는 과정을 연구하였다. Eriksson 등[28]은 peroxisome proliferator-activated receptor-gamma 2 (PPAR-γ2) 유전자의 Pro12Ala polymorphism이 적정체중에서와는 달리 부당경량아에서 인슐린 저항성을 증가시킨다고 하였다. 만약 태아에게 저체중을 유발하면서 인슐린 저항성을 함께 유발하는 유전자가 있다면 그 기전을 이해하는데 용이하겠지만, 현재까지 이러한 유전자는 알려진 바가 없다. 다만, Hattersley 등[29]에 의하면 glucokinase에 돌연변이가 있는 영아들이 동시에 출생체중이 작은 것을 관찰하여 이후에 당뇨로 발병할 가능성이 있음을 제시하였으나 glucokinase 돌연변이가 매우 희귀하기 때문에 일반적으로 저체중과 2형 당뇨의 연관성을 설명하기에는 설득력이 떨어진다. 이상을 종합해 볼 때, 인슐린 저항성의 획득에는 산모의 영양 및 환경적 요인과 유전자적 요인이 모두 복합적으로 관여할 것으로 여겨지며 결국, 위에서 설명한 절약형질의 발현, 절약 유전자형의 역할이 모두 관여하는 것으로 설명할 수 있다(Figure 1).
성장과정에서 대사증후군으로의 이환
이러한 위험성이 있는 영아들은 성장과정에서 지속적으로 대사증후군으로 이행하고 있는 것으로 여겨진다. Ortiz-Espejo 등[30]은 1,500 g 미만으로 출생하여 교정 연령 36주에 체중이 3백분위 미만에 머문 자궁외 성장부전(extra-uterine growth restriction, EUGR)에 해당했던 미숙아 38명과, 건강하게 만삭으로 출생했던 123명의 대조군을 대상으로 사춘기 전(prepubertal)에 채취한 adiponectin, resistin 등의 혈중 농도를 비교하였다. 이 연구에서 EUGR을 보였던 사춘기 전 아이들은 그렇지 않았던 아이들에 비해서 유의하게 adiponectin이 낮고 resistin은 높아 대사증후군에서 나타나는 지방세포 및 염증 유발 사이토카인의 특성을 보였다. 자궁 내 성장 부전 미숙아에서 adiponectin이나 resistin의 농도에 대한 연구결과는 높다는 보고와 낮다는 보고가 공존하지만, Ortiz-Espejo 등[30]의 연구는 EUGR의 병력이 있는 아이들을 대상으로 사춘기 전기에 비교 분석한 유일한 연구로서 결국에는 청소년 전기에 이르러 대사증후군처럼 낮은 adiponectine과 높은 resistin을 나타내는 것을 증명하였다. 태내에서 자궁 내 성장 부전을 보였던 만삭아를 대상으로 하였던 다른 연구들과는 달리 이 연구에서는 조기분만이 되지 않았더라면 성장부전에 노출되지 않았을 수도 있는 미숙아들을 대상으로 하였다. 이 연구에서 흥미로운 점은 대상환자들이 교정연령 36주에 성장부진을 보였지만, 미숙아 치료를 하는 동안에 대부분의 신생아학 의사들이 영양을 과잉으로 공급했다는 사실이다. 실제로 미숙아의 치료의 과정에서 총정맥 영양뿐만 아니라 장관 영양에 있어서도 모유강화제나 미숙아 분유를 사용하여 120-130 kcal/kg의 고열량을 공급하고 있다. 왜냐하면 극소 저체중 출생아(출생체중 1,500 g 미만) 및 초극소 저체중 출생아(출생체중 1,000 g 미만)의 환자들이 하나 혹은 그 이상의 동반질환을 가질 수록 EUGR이 심하고 이러한 경우에 신경발달학적 결과 및 동반 질환의 예후가 나쁘기 때문이다[31].
따라잡기 성장과 지방 형성 반동
위에서 언급한 영양학적 치료 전략은 미숙아들이 퇴원 이후에도 연장되어 '따라잡기 성장(catch-up growth)'을 유도하게 된다. 부당 경량아의 경우 대개 1세 이전에, 77.1%에서는 2세까지 따라잡기 성장을 완성한다. 그러나 이른 시기에 따라잡기 성장이 급격하게 이루어진 경우 대사증후군으로의 진행은 더욱 심화된다. 동물실험 연구를 예로 들면, 임신한 어미에게 영양을 제한하여 자궁 내 성장 부전을 유도한 후에 새끼로 하여금 과식으로 조기에 따라잡기 성장을 시킨 경우 새끼 동물들은 더욱 비만하고 지방조직의 비율이 클 뿐만 아니라 유리 지방산의 인슐린에 대한 감수성도 증가하였다[32]. 사람에서도 동일한 현상을 관찰할 수 있는데, 저체중으로 출생하여 영아기나 소아기에 따라잡기 성장을 한 군에서 그렇지 않은 군에 비해 비만과 대사증후군으로 발전하는 위험도가 더 높았다[33]. Euser 등[34]은 32주 미만으로 출생한 403명을 전향적으로 추적관찰 한 연구에서 출생 초기 및 영아기에 따라잡기를 많이 한 아이들일 수록 19세때 체질량 지수, 체지방율, 그리고 복부지방 등이 많이 증가한 것을 보고하였다.
태아기에서 이른 영아기까지 동화작용(anabolism)에 가장 중요한 호르몬은 인슐린이다. 즉, 인슐린이 촉매제로 작용하여 포도당을 포획하고 지방합성이 진행된다. 따라서 인슐린 자체 혹은 저항성에 문제가 생기면 지방조직이 증가하거나 유리 지방산이 증가하는 등의 이상이 생기고 이상지질혈증, 비만, 당뇨 등의 대사증후군의 위험이 함께 커지게 된다. 지방세포가 지방 전구세포로부터(preadipocyte) 분화하는 데에는 영양이나 호르몬 외에도 PPAR-γ, sterol regulatory element binding protein-1 (SREBP-1) 등의 전사인자(transcription factor)가 촉매작용을 한다. EUGR이 있는 미숙아들에서 지방합성이 과장되어 발현되는 이유에 대해서 이러한 환자군에서 PPAR-γ, SREBP-1 등이 증가되어 있다는 것이 최근에 여러 연구들을 통하여 밝혀졌다. 따라서 이러한 소인이 있는 성장지연이 있는 환자에서 빠른 따라잡기 성장이 비만세포의 과증식을 일으킬 수 밖에 없는 것이다[3536].
체질량 지수의 급속한 증가는 대부분에서 생후 첫해, 즉 영아기에 이루어진다. 정상적으로 이러한 증가 속도는 점차 감소하여 생후 6세 경에 이르면 가장 낮은 속도를 나타내다가 다시 증가하게 되는 시기에 이르는데 이러한 때를 체지방 형성 반동(adiposity rebound)이라고 한다[37]. 체지방 형성 반동에 이르는 시기가 빠르면 빠를수록 대사증후군에 잘 이환된다고 알려져 있다. 한편, 인도에서 시행한 몇몇 연구에서 대사증후군이 체중 자체의 문제라기보다는 오히려 비정상적 체조직의 구성(body composition)에 기인하는 것을 시사하였다. 한 연구에 의하면 유럽과 비교하여 산모의 영양이 불균형적이었던 산모들의 아기들이 평균적으로 800 g 정도 작았던 것이 지방 뺀 체중(lean body mass=body weight–body fat)이 작았기 때문이고 오히려 복부지방(abdominal adiposity) 비율은 크게 나타났다. 즉, 태아 및 신생아에 있어서도 체중은 작고 체지방은 높은 마른 비만(thin-fat baby)이 가능하여 2형 당뇨에 이환될 확률이 높다는 것이다[38].
저체중 출생아, 미숙아에서 신경 발달학적 저하, 인지기능 및 학습기능의 지연 등을 우려하여 고칼로리의 영양을 공급하고 있으나, 최근에 이러한 전략이 과연 이득이 있는가에 대한 분석이 이루어지고 있다. Young 등[39]이 2013년에 발표한 메타분석에 의하면 2개의 연구에서 총 246명의 영아를 분석해 보았을 때, 퇴원 시 수유로 모유강화제를 사용한 군에서 성장 및 발달에서 특별한 이득이 없었다. 따라서 저체중으로 출생한 영아, 특히 미숙아에서 EUGR을 보인 영아에 있어서 전통적으로 시행해온 것처럼 고영양 식이를 공급하여 따라잡기 성장을 서두를 필요가 없으며, 오히려 위에서 고찰해 보았듯이 성인이 되었을 때, 인슐린 저항성과 당뇨, 비만을 포함하여 대사증후군으로 이환될 위험이 매우 크므로 급속한 따라잡기 성장은 지양해야 할 것으로 생각한다. 이러한 의미에서 미숙아에서도 최소한 6개월까지 완전 모유 수유를 권장하는 것이 하나의 대안이 될 수 있다[18].
그러나 Bocca-Tjeertes 등[40]에 의하면 출생체중이 작은 미숙아는 출생체중이 적절한 미숙아 및 만삭아보다 따라잡기 성장이 느리고 심하면 생후 5세까지 늦어지는 것으로 보고되었고, 신경 발달학적으로 예후가 떨어지는 것이 사실이기 때문에, 영양학적 지지를 언제까지 할 것인가에 대한 연구는 더 이루어져야 할 것이다.
결론
비만과 대사증후군은 소아에서도 중요한 질환군의 하나이며 발병 기전에는 여러 가지가 있지만 그 기원을 태아 및 신생아기에서부터 절약 유전자형 및 절약 형질의 발현으로 설명할 수 있다. 이러한 변화들은 태내에서 인슐린 저항성을 나타나게 하고 이후 따라잡기 성장을 하는 과정에서 영아기 및 소아 청소년기 전반에 걸쳐 대사증후군으로 연결된다. 특히 자궁 내 성장지연 및 저체중 출생아, 그리고 신생아 중환자실 치료과정에서 발육부전이 심화된 극소 저체중 출생아 등이 고위험군이다. 따라서 이러한 환자군일수록 영아기에 따라잡기 성장을 서두르지 않도록 하고 소아기 및 청소년기에도 대사증후군에 대한 고위험군으로서 특별한 관리가 필요하다.
Peer Reviewers' Commentary
비만과 대사증후군은 소아에서도 중요한 질환군의 하나이며, 소아의 대사증후군은 성인에서도 지속되어 국민 건강학적 측면에서 큰 문제로 대두되어 있다. 본 연구에서는 현재까지 알려진 발병 기전들에 대하여 다각도로 분석하여 설명하였다. 특히 자궁내 성장 지연 및 극소 저체중 출생아 등의 고위험군에 대하여 현재 시행하고 있는 치료 원칙인 고영양 공급이 대사증후군을 야기할 수 있을 것으로 추정하였다. 따라서 이러한 환자군일 수록 영아기에 따라잡기 성장을 서두르지 않도록 하고 소아기 및 청소년기에도 대사증후군에 대한 고위험군으로서 특별한 관리가 필요하다고 결론하고 있다.
[정리: 편집위원회]
 XML Download
XML Download