

 PDF
PDF ePub
ePub Citation
Citation Print
Print


서론
우리나라는 지금까지 약물 남용의 안전지대로 여겨졌으나 최근 약물 남용 사례가 급증하고 있고 약물 남용 계층도 점차 저연령화, 다양화되고 있어 심각한 사회문제가 되고 있다. 그리고 과학기술이 발전함에 따라 수많은 새로운 화학물질이 의약품으로 도입되어 신종 마약류가 계속 출현할 뿐만 아니라 사회가 복잡해지고 다양화됨에 따라 이러한 약물의 사용이 향후 증가할 것으로 예상된다. 약물 남용에 대한 효과적인 치료가 이루어지기 위해서는 약물이 뇌에 미치는 작용 메커니즘 및 남용 약물의 특성, 물리화학적 성질 및 약동 및 약력학적 특성에 대한 포괄적인 이해가 필요하다. 이 특집에서는 약물 남용에 의해 발생하는 신경생화학적 기전 및 남용 약물들의 약동 및 약력학적 개요 및 각 약물의 특성을 살펴보고자 한다.
약물 오남용의 신경생물학적 기전
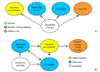
뇌에서 중격의지핵(nucleus accumbens), 편도핵(amygdala) 및 전전두엽피질(prefrontal cortex)은 적응 행동(adaptive behavior)을 관여하고 조절하는 주요 부위이다. 중변연계(mesolimbic system)의 앞 부위에 위치하는 중격의지핵은 양성보상행동(positive reward behavior)에 관여하고, 편도핵은 음성 혹은 공포동기화행동(negative or fear-motivated behavior)을 매개하며, 전전두엽피질은 보상행동에 대한 예측 및 의사결정에 관여한다[1]. 자연 보상(natural reward) 행동(예: 성욕, 식욕) 및 약물 남용의 경우 뇌에서는 복측피개부(ventral tegmental area)에서 중격의지핵을 연결하는 경로를 통해 중격의지핵에서의 도파민(dopamine) 양이 증가한다. 코카인, 암페타민, 메틸렌디옥시메스암페타민(methylenedioxymethamphetamine) 등은 직접 도파민 재흡수를 방해하거나 분비를 증가시키고, 알코올, 아편유사제, 니코틴(nicotine), 대마(cannabis), NMDA 길항제 등은 도파민 세포의 발화를 조절하는 신경세포를 자극함으로써 간접적으로 도파민 양을 증가시킨다[2]. 초기 약물 남용 단계에서는 중격의지핵에서의 도파민 증가가 주요한 역할을 한다. 그 외 GABA, 아편유사제 펩타이드(opioid peptide), 세로토닌(serotonin), 아세틸콜린(acetylcholine), 엔도카나비노이드(endocannabinoid) 및 글루타메이트(glutamate) 등이 직접 혹은 간접적으로 복측피개부 및 중격의지핵에서의 도파민 분비에 영향을 미친다. 이 중 GABA 사이신경세포(interneuron)는 복측피개부와 중격의지핵에서의 도파민 분비를 억제시키는 작용을 하며, 아편유사제는 μ수용체에 작용하여 GABA 사이신경세포 작용을 억제시킴으로써 도파민 전달을 활성화시킨다[34]. 중격의지핵에서의 도파민 증가는 자연 보상 시에 비해 약물 남용 시에 훨씬 급격하게 나타나며, 그 증가량도 많다[5]. 또한 약물 남용은 학습과 기억 영역의 신경생물학적 기능을 변화시켜 적응 불량 상태를 학습시킴으로써 발생하는 일종의 기억 질환(memory disorder)이라는 주장도 있다[6].
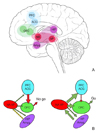
약물 남용 초기 단계에서 약물에 의존적으로 전환되어 가는 과정에서 약물들은 뇌의 스트레스 시스템을 자극하게 되며, 금단 시 편도핵에서 부신피질자극호르몬(adrenocorticotropic hormone), 콜티코스테론(corticosterone), 부신피질 자극호르몬 방출인자(corticotrophin releasing factor) 등이 증가한다. 이러한 뇌의 스트레스 시스템의 활성화는 금단 증상이나 부정강화(negative reinforcement)를 유발하며, 부신피질 자극호르몬 방출인자 길항제와 같은 항스트레스 약물은 향후 새로운 약물 남용 치료제로 사용될 것으로 기대된다[7]. 초기 약물 남용 단계의 보상행동 시, 관찰되던 도파민 중심의 신경체계는 점차 강박적이고 억제되지 않는 약물 중독 단계로 진행되면서 글루타메이트 중심의 체계로 변화하게 된다[38]. 초기에는 중격의지핵에서 도파민이 분비되지만, 남용이 점차 진행됨에 따라 도파민 분비가 전전두엽피질과 편도핵에서 이루어진다. 이렇게 분비된 도파민은 전전두엽피질과 편도핵 사이의 글루타메이트 전달을 촉진하고 또한 전전두엽피질에서 중격의지핵 중핵(core)으로 글루타메이트분비를 증가시킨다[89] (Figure 1). 중독 상태에서 반복적인 약물 투여가 지속되면 결국 전전두엽피질에서 중격의지핵으로의 글루타메이트 분비 조절 기능이 상실되고, 중격의지핵의 글루타메이트 세포외 기초량(basal extracellular level)은 감소하게 된다. 전전두엽피질 기능의 상실은 자제력 상실, 충동성, 부적절한 의사결정 등을 야기한다. 앞띠이랑(anterior cingulate gyrus)은 행동 통제 기능을 담당하는데 앞띠이랑의 활동도가 감소하면 약물 갈망에 대한 자제력을 잃게 된다. 눈확이마피질(orbitofrontal cortex)는 외부의 물리적 요소와 정서적 요소를 통합하여 동기부여를 지정하는 역할을 하는데 손상 시 약물 갈망 및 부적절한 의사결정을 초래하게 된다[210] (Figure 2).
남용 약물의 약동, 약력학적 개요
1. 약물의 흡수
물리화학적 성질, 조제 형태(dosage form), 투여 경로, 일차 통과 효과(first pass effect), 전구약물(prodrug) 여부에 따라 약물의 흡수(absorption) 및 생체이용률(bioabailability)이 결정되며, 이는 약물의 작용 발현시간에 영향을 미친다. 지질 용해도(lipid solubility)가 큰 약물일 수록 빨리 흡수되며 생체이용률도 증가하게 되어 남용의 위험이 커진다[11]. 동일 성분의 약제라도 화학적 형태(chemical form)에 따라 자가 투여(self-administration) 방법이 달라지는데, 코카인(cocaine)이 대표적인 예이다. 입자의 크기, 가용성, 첨가제(excipient) 및 분비속도(rapid or slow-release) 등의 조제 형태에 따라 약물 남용의 위험성도 달라진다. 남용의 위험도를 줄이기 위해서는 원래의 제형을 변경시키기 힘들도록 제조하는 것이 중요한데, 예를 들어 경구용 마약류를 부수어 정주용으로 사용하는 경우 약물에 첨가제를 넣어 녹지 않도록 만들어 줌으로써 남용의 위험도를 줄일 수 있다[12]. 투여 경로에 따라서도 약물의 흡수 및 발현 시간은 달라지는데, 정주 및 흡연 시 약물의 흡수 및 발현 시간이 빠르다. 정주에 비해 흡연 시의 흡수 및 발현 시간이 빠르고 효과도 강력한데, 이는 폐를 통한 약물의 흡수가 빠르고, 폐혈관에서 뇌로의 약물 전달이 빨리 일어나기 때문이다. 약물의 경구투여는 상대적으로 남용의 위험성이 낮다[13].
2. 약물의 분포
약물이 흡수되면 혈관 내를 순환하며 여러 신체 조직으로 분포한다. 남용되는 대부분 약물의 효과처는 뇌조직이므로 약물이 이 곳으로 분포되어야만 효과를 나타낸다. 약물이 혈액에서 뇌로 이동하여 효과를 나타내는 데는 물리화학적 성질(지질 용해도, 분자크기, pH, pKa 등), 약물 운반 형태(passive diffusion or active transport) 및 혈류량 등이 영향을 미치게 된다. 예로서 지질 용해도가 큰 바르비투르산염(barbiturate)나 벤조다이아제핀은 약물 작용 부위인 뇌로의 이동이 빠르게 일어나며, 지질 용해도가 큰 약물이 남용의 위험도가 상대적으로 크다[11].
3. 약물의 제거
약물의 생화화적 변형은 주로 간에서 이루어지며, 그 외 폐, 신장 등에서도 일부 이루어진다. 시토크롬(cytochrome) P-450은 간 대사에서 대표적인 마이크로솜(microsome) 효소 시스템이다. 마이크로솜 효소 시스템은 페노바르비탈(phenobarbital), 리팜핀(rifampin), 알코올 혹은 담배 등에 의해 활성화가 되기도 하고, 페니토인(phenytoin), 클로람페니콜(chloramphenicol) 혹은 시메티딘(cimetidine) 등에 의해 활성이 저하되기도 한다[14]. 경구투여 후에 일차 통과 효과를 거친 약물의 대사산물이 활성 물질이 되는 경우는 남용의 위험이 크다. 약물 및 대사 산물의 대부분은 신장을 통해 배설되며, 그 외 대변이나 호흡을 통해 배설되기도 한다[11].
남용 약물의 약동학과 약력학
1. 아편유사제
1) 모르핀
경구투여 후 일차 통과 효과가 커서 생체이용률은 약 30% 정도이고, 2시간의 반감기를 가지며 30%가 단백결합을 한다. 대사는 글루쿠론산(glucuronic acid)과 포합반응을 하여 morphine-3-glucuronide (M3G)와 morphine-6-glucuronide (M6G)와 같은 대사산물이 생성된다. M3G는 비활성대사산물인데 반해 M6G는 활성대사산물로서 진통효과를 나타낸다. M6G는 모르핀보다 긴 반감기를 가지므로 신장질환 환자의 경우 M6G가 축적되어 호흡저하 등을 초래할 수 있다[16].
2) 헤로인
진통효과는 모르핀의 2배이며, 전구약물로서 헤로인(heroin) 자체의 약리효과는 미미하다. 가수분해를 통해 6-monoacetylmorphine (MAM)으로 빠르게 대사된 후 모르핀으로 전환된다. MAM은 지질 용해도가 커 중추신경계로 신속하게 이동하고 이로 인한 진통효과가 나타나며, 반감기는 짧다. 헤로인 염기(base)나 알칼로이드(alkaloid)는 녹는 점이 낮아서 가열을 이용한 증기 형태로 남용되며, 흡입 시 중추신경계로 신속하게 이동한다. 카페인(caffeine)이나 페노바르비탈(phenobarbital)과 같은 희석제(diluent)를 사용하는 경우 생체이용률이 증가한다. 헤로인 염기에 레몬 쥬스를 첨가한 염(salt)제재가 정주용으로 남용되기도 한다[17].
2. 마약 중독 치료약제
1) 메사돈
메사돈(methadone)이 중추신경계에 미치는 영향은 모르핀과 비슷하고 진통작용의 강도 및 지속시간도 거의 같다. 합성 아편계 약물로서 주로 모르핀이나 헤로인 의존성이 생긴 환자의 금단증상 치료에 사용된다. 우선성(d) 광학이성체(enantiomer)는 16시간의 반감기를 가지고, 좌선성(l) 광학이성체는 48시간의 반감기를 가지며, 좌선성 광학이성체가 우선성 광학이성체보다 진통효과는 8-50배 강하다. 경구투여 시 흡수가 잘 되어 4시간 후 혈중 최고치에 도달하고 80% 이상의 생체이용률을 나타내며, 피하주사 시는 10분만에 혈중에 나타난다. 3구획 약동학 모델에 적합하며 지질 용해도가 커 중추신경계로의 이동이 빠르다. 약동학적 개인차가 크며 주로 제거 반감기(13-58시간) 및 청소율에서 큰 개인간 변이를 나타낸다. 높은 단백결합률(85-94%)과 큰 분포용적(3.6 L/kg)을 가진다. 주로 간에서 N-demethylation에 의해 대사되어 비활성 대사산물인 pyrrolidine이 생성되며, 이는 소변이나 담즙으로 배설된다. 약동학적 개인차가 커 예상치 않은 호흡 억제가 발생할 수 있고 치료 시 소용량(20-40 mg)부터 시작하여 4주 이상에 걸쳐 단계적으로 증량한 후에 지속 용량(50-120 mg/d)을 사용할 것을 추천한다[1819].
2) 날트렉손
날록손(naloxone)의 화학적 변형 제재로 아편유사제 길항제(antagonist)이다. 경구투여 후 80%가 흡수되나 일차통과 효과가 커서 경구투여 용량의 5%만이 혈장내로 들어온다. 20%의 단백결합률을 나타내며 분포용적이 크다. 작용 지속시간은 날트렉손(naltrexone) 자체의 효과보다는 주요 대사산물인 β-naltrexol (6-β-hydroxynaltrexone)에 의해 좌우되며, β-naltrexol의 반감기는 20시간이다. 날트렉손의 마약 차단 효과의 작용 지속시간은 용량 의존적이며, 마약 수용체에 대한 친화력은 헤로인의 100배 정도이다. 대량을 장기간 사용하여도 의존성은 잘 생기지 않으며 투약을 중지해도 금단증상이 잘 나타나지 않는다[20].
3) 부프레노르핀
μ수용체 부분작용제(partial agonist)로서 지질 용해도가 크다. 경구투여 시 생체이용률은 15%이고, 설하투여 시는 55%이며, 설하투여 시 흡수가 빠르고 중추신경계 내로의 이동이 빨라서 설하투여가 선호된다. 간에서 대사되어 대사산물인 N-dealkylbuprenorphine이 생성되며 배설은 주로 담즙과 대변을 통해 이루어진다. 설하투여 시 진통효과는 투여 후 15-45분 정도면 나타나고 8시간 이상의 작용 지속시간을 나타낸다[21]. 부프레노르핀(buprenorphine)은 수용체와의 해리가 매우 느리게 일어나므로 저용량에서는 금단증상이 약하게 나타나고 고용량에서는 긍정강화(positive reinforcement)를 감소시킬 수 있는 장점이 있다. 약물치료에 대한 순응도가 좋으며 고농도에도 호흡억제가 드물어 메사돈에 비해 안전하다[22].
4) 클로니딘
클로니딘(clonidine)은 중추와 말초의 α2-아드레날린 수용체 작용제로서 마약, 알코올, 니코틴 등의 정신작용제(psychoactive drug)의 금단증상 치료에 사용된다. 금단증상 시 교감신경 활성으로 인해 발생하는 고혈압, 빈맥, 전율(tremulousness), 복부경련 및 약물 갈망현상을 감소시킨다. 경구투여 시 흡수율이 뛰어나 생체이용률이 100%에 가깝고, 약물의 절반 가량은 간에서 대사되어 비활성대사물인 p-hydroxyclonidine과 부분활성대사물인 2,6-dichlorophenylguanidine이 생성되며, 나머지 절반은 대사과정을 거치지 않고 소변으로 배설된다. 반감기는 12시간 정도이며 개인간 변이가 크다. 경구투여 시 2-3시간에 혈장 농도가 최대에 이르며, 이 때 항고혈압 작용도 최고의 효과를 나타낸다[2324].
3. 중추신경 흥분제
1) 코카인
코카인은 코카나무 잎 중에 함유되어 있는 알칼로이드로서 중추신경 흥분 효능을 갖고 있다. 지용성이고 약알칼리성(pKa 8.6)이며, 화학적 형태에 따라 코카인 염산염(hydrochloride; 'snow', 'coke')과 코카인 자유염기(freebase; 'crack', 'rocks')의 두 종류로 나뉜다. 경구, 코로의 흡입, 정주 및 흡연 등 다양한 경로로 투여된다. 코카인 염산염은 미세분말 형태로 코로의 흡입이나 정주로 사용되며, 코카인 자유염기는 열에 잘 분해되지 않고 휘발성이 있어 흡연을 통해 투여된다. 흡연 시에는 증기(6%) 및 에어로졸 입자(94%)가 혼합된 형태로 전달되며, 폐에서의 넓은 흡수 면적 때문에 중추신경계로 전달되는데 걸리는 시간은 6-8초 정도이다. 정주투여 시에는 중추신경계 효과가 12-16초 후에 나타나는 반면 코 흡입 시에는 2-5분 후에 나타난다. 코 흡입 시에는 코카인 자체의 혈관수축 효과로 인해 흡수가 저해되고 생체이용률은 20-60% 정도이다. 만성적으로 사용 시에는 코 점막이 무혈관 상태로 변해 흡수율이 감소한다. 90%가 단백결합을 하며, 향정 상태(steady-state) 시의 분포면적은 1.96 L/kg이다[25]. 정주 시 2구획 모델에 적합하며 분포단계(distribution phase)는 매우 빠르고 제거 반감기는 31-82분 정도이다.
대사는 3가지 경로를 통해 일어나는데 혈장 에스트라제(esterase), 가성콜린에스트라제(pseudocholinesterase) 및 간 에스트라제(hepatic esterase)에 의해 ecgonine으로 대사되고(32-49%), 가수분해 및 간 카복시에스트라제(hepatic carboxyesterase)에 의해 benzoylecgonine으로 대사되며(35-45%), CYP3A에 의해 norcocaine으로 대사된다(2.6-6.2%). 여자가 남자에 비해 대사속도가 빠른데 이는 여자의 적혈구 콜린에스트라제(cholinesterase) 활성도가 더 크기 때문이다. 또한 남자에게서 혈중 최고 농도에 도달하는 시간이 빠르고 최고 농도도 높으며 작용 지속시간도 더 길다. 코카인 복용자의 50-90%는 알코올을 함께 복용하며, 이 때의 알코올은 간 카복시에스트라제 대사를 감소시켜 대사산물인 benzoylecgonine의 생성이 감소되고, transesterification에 의해 대사산물인 코카에틸렌(cocaethylene)이 생성된다. 이렇게 생성된 코카에틸렌은 도파민 재흡수 펌프(reuptake pump)를 억제하여 코카인과 함께 도취감(euphoria)을 증강시킨다. 코카에틸렌은 제거반감기가 3.5-5.5시간으로 코카인보다 길며 축적 작용이 있다.
카테콜아민(catecholamine)을 연접틈새(synaptic cleft)로 분비시키고 아민(amine) 신경전달물질의 재흡수를 방해하며 신경세포에서의 나트륨 통로를 차단한다. 도취감(euphoria)에 대한 내성이 빨리 발생하므로 고용량의 코카인을 찾거나 투여 횟수를 늘이게 되며, 이러한 경우 환시(visual hallucination), 편집(paranoid) 및 우울증 등과 같은 급성 기질적 뇌증후군(organic brain syndrome)이 발생할 수 있다. 고용량 투여 시는 중추신경계 과흥분, 빈맥, 고열증, 동공확대, 혈관수축 등의 교감신경 항진 증상이 나타나며 장기간 사용 시 여자에서 무월경 및 불임과 같은 내분비적 장애가 발생한다. 코카인 중독 치료 약제로는 항우울제, 도파민 작용제, 도파민 길항제, 아편유사제, 항경련제 등이 사용된다[26].
4. 환각제
1) 암페타민
중추신경계에 강력한 흥분작용을 일으키는 교감신경 유사제로서 식욕 억제, 각성 유발 및 들뜬 기분을 유발한다. 삼키거나 코로의 흡입, 정주 및 흡연 등 다양한 경로로 투여된다. 경구투여 시 15-30분에 효과가 나타나기 시작하여 1-1.5시간에 혈중 최고 농도를 나타내고, 흡연 시에는 혈중 최고 농도 도달 시간이 더 빨라진다. 단백결합률이 낮고(20%) 반감기는 12-36시간인데 정상인에 비해 남용자의 반감기가 더 길다. 또한 암페타민(amphetamine) 남용자(6.1 L/kg)는 정상인(3.5-4.6 L/kg)에 비해 분포용적이 더 큰데, 이는 반복사용에 따른 약물의 조직에 대한 친화력이 증가하여 나타나는 현상이다. 암페타민은 알칼리성(pKa 9.9)이므로 소변을 산성화시키면 신장으로의 배설이 촉진되는데, 급성중독 시 염화암모늄(ammonium chloride) 등을 투여하여 약물의 배설을 촉진시키기도 한다. 중독자들은 약물의 작용시간을 늘이기 위해 중탄산염(bicarbonate)제산제를 섭취하여 소변을 알칼리화함으로써 작용시간을 늘이기도 한다. 남용 시 발생하는 정신병적 증상은 암페타민혈중 농도보다는 소변으로 배설되는 극성 대사산물(norephedrine, p-hydroxynorephedrine, hydroxyamphetamine)의 농도에 비례한다. 신경전달물질의 분비를 촉진시키고 모노아민(monoamine) 신경전달물질의 흡수를 방해함으로써 결국 모노아민 고갈과 수용체 민감성을 초래한다[27].
메스암페타민(methamphetamine)은 암페타민의 N-methyl 유도체이다. 수면제 과다복용으로 인한 호흡곤란 환자에서 호흡 촉진을 위해 사용되며, 과거 비만환자의 치료목적으로도 사용되었다. 현재 암페타민 약물 계열 중에서 남용 사례가 가장 심각하다. 약력학적 작용은 암페타민과 비슷하나 혈액뇌장벽(blood-brain barrier) 통과가 더 용이하므로 중추신경 작용이 강하며 증강된 약효를 나타낸다. 주로 중추신경 흥분작용만 강하고 말초 작용은 별로 없다. 그 외에 유사체들로 3,4-methylenedioxymetamphetamine (MDMA; Ecstacy), 3,4-methylenedioxyamphetamine (Eve), 2,5-dimethosy-4-methylamphetamine, p-hydroxydimethoxy-4-methylamphetamine 등이 있다. 암페타민이나 MDMA를 모노아민 옥시다제 억제제(monoamine oxidase inhibitor)인 페넬진(phenelzine)이나 트라닐사이프로민(tranylcypromine) 등과 혼합 투여 시 약력학적 상호작용에 의해 고혈압, 빈맥, 혼돈(confusion), 혼수(coma) 및 심한 경우 사망에 이를 수 있다[28].
2) 대마
주 성분은 delta-9-tetrahydrocannabinol (THC)이며, 동일 효력을 나타내기 위해서 흡연으로 투여하는 것이 경구투여보다 효과적이다. THC는 지질 용해도가 커서 분포용적이 약 500 L에 달한다. 흡연 시 혈액으로 신속하게 흡수되어 다양한 조직으로 분포되는데, 제일 먼저 뇌로 도달하고 30분 이내에 뇌에서 사라진다. 5-10분 이내에 중추신경계 효과와 심혈관계 효과가 동시에 나타난다. 개인 간의 흡연 패턴에 따라 혈액 흡수 정도가 다른데, 남용자의 경우 혈액 흡수율이 높다. THC의 반감기는 19시간 정도이고, 주 대사산물인 11-hydroxy-delta-9-THC의 반감기는 50시간이다. THC를 경구투여하면 흡수가 느리고 간에서 11-hydroxy-delta-9-THC로 변하므로 뇌에 도달하는 THC 양은 적고, 중추신경계 효과가 나타나는데 많은 시간이 걸린다. 진정 및 진통작용을 갖고 있으나, 다량 사용 시 환각효과를 나타낸다. 다른 약물에 비해 의존 가능성이 상대적으로 낮은 것으로 알려져 소수의 사람들이 위험성을 간과하는 경향이 있다. 심박수를 증가시키고 사람에 따라서는 혈압을 상승시킨다[29].
5. 중추신경억제제
1) 벤조다이아제핀
벤조다이아제핀(benzodiazepine) 계통의 약물들은 대부분 지질 용해도가 크며, 경구 투여 시 지질 용해도가 더 큰 약물일수록 위장관내에서의 흡수 속도가 빨라 약물의 효과도 빨리 나타나므로 남용의 위험도가 커진다. 예로서 다이아제팜(diazepam) 및 알프라졸람(alprazolam)이 지질 용해도가 상대적으로 작은 할라제팜(halazepam) 및 옥사제팜(oxazepam)보다 남용 위험도가 크다[11]. 중추신경계 효과를 신속하게 할 목적으로 경구용 벤조다이아제핀을 녹여 정주용으로 사용하기도 한다. 다이아제팜이나 미다졸람(midazolam)과 같이 지질 용해도가 큰 약물인 경우는 혈장에서 중추신경계로의 이동 속도 반감기는 1분 이내이다. 다이아제팜과 활성대사산물인 desmethyldiazepam (nordazepam)는 제거반감기(elimination halflife)는 길지만, 뇌, 폐 및 지방으로의 분포가 신속히 일어나 효과 지속 시간은 그리 길지 않다. 반복투여 시 체내 축적이 일어나며, 이는 근육량에 비해 지방량이 많은 노인 환자의 경우 심하다. 분포 용적은 1 L/kg 내외이다. 금단 증상의 발생과 심한 정도는 반감기와 관계가 있으며, 옥사제팜과 같이 짧은 반감기(1-3일)를 가지는 약물은 금단증상의 발생이 빠를뿐더러 심하게 나타나는데 반해 긴 반감기(4-8일)를 가지는 약물의 경우 증상이 늦게 나타나며 그 증상도 심하지 않다[30]. 용량 의존적으로 불안해소, 진정 및 호흡 억제가 발생하며, 기억상실과 조화운동불능(incoordination) 등이 나타날 수도 있다. 벤조다이아제핀 의존성은 변연계의 GABA-벤조다이아제핀 수용체의 하향조절(downregulation)과 역 작용제(inverse agonist)에 대한 민감성 증가와 상관이 있다. 벤조다이아제핀 급성중독의 치료제로 주로 사용되는 플루마제닐(flumazenil)은 경구로의 흡수가 잘 이루어지지 않기 때문에 정주용으로 주로 사용되며 의존성 치료제로는 한계가 있다[31].
결론
최근 불법적으로 유통되는 약물은 식욕 억제, 환각 및 각성의 목적으로 그 종류와 사용 사례가 날로 증가하고 있다. 또한 제품의 형태도 정제 또는 주사제와 같은 의약품뿐만 아니라 건강식품 등에 이러한 약물을 불법적으로 첨가하여 유통하는 사례도 자주 적발되고 있다. 그 효과가 강력할 뿐 만 아니라 부작용을 예측하기가 어려워 사용자의 피해가 매우 크다. 그리고 과학기술이 발전함에 따라 신종 마약류가 계속 출현할 뿐만 아니라 사회가 복잡해지고 다양화됨에 따라 이러한 약물의 사용이 향후 증가할 것으로 예상된다. 약물 남용 확산방지를 위한 범정부차원의 교육 및 홍보가 절실하며, 이와 함께 약물치료법의 근간이 될 수 있는 신경생물학적 기전 및 약물의 약동, 약력학에 대한 지속적인 이해 및 연구가 필요할 것이다.
Peer Reviewers' Commentary
본 논문은 마약, 환각제, 중추신경 흥분제 및 억제제들이 어떤 기전으로 중독 및 오남용을 일으키는 지에 관해 일목요연하게 기술하고 있다. 중독 및 오남용을 유발시키는 약물들이 중격의지핵(nucleus accumbens)에서의 도파민 분비 증가와 이로 인한 보상감과 쾌감, 이후 뇌의 스트레스 시스템의 활성화, 그리고 마지막으로 도파민에서 글루타메이트 중심의 체계로의 전환이 이루어지면서 정상적인 뇌기능을 상실하게 되는 공통 경로를 밟는다는 점은 매우 흥미롭다. 이러한 기전의 이해는 약물 중독 및 오남용을 치료하고 예방하는 데 있어 의료인이 반드시 숙지하고 있어야 할 필수적인 사항이다. 본 논문은 의료인으로 하여금 그러한 약물의 중독 및 오남용 기전을 쉽게 이해할 수 있도록 설명하고 있다.
[정리: 편집위원회]

 XML Download
XML Download