

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Transmissible Spongiform Encephalopathy (TSE) or prion diseases are fatal neurodegenerative diseases, which are caused by transmissible abnormal prion proteins, converting the endogenous normal prion in the body to the infectious abnormal prions. The most common form of human prion diseases is Creutzfeldt - Jakob disease (CJD). Most of CJD are sporadic with unknown cause. Some familial or iatrogenic CJDs are reported in many countries, but there have been no formally reported case in Korea. Variant CJD (vCJD) is a new form of human prion disease, which revealed differentiated clinical presentations and laboratory diagnostic results. vCJD was thought to be originated from eating the beefs or other parts of bovine spongiform encephalopathy (BSE) infected cattle. The unpredictable species barriers, the underestimated distribution of prion infected tissues, the variable clinical courses, and uncertain disease progressions of many prion diseases, all made the prion related risk assessment very difficult. Korea needs our own surveillance system for various prion diseases of human and animals and to make plans for the risk assessment of the various prion disease transmissions for the minimal spread by maximizing the research capacities.
Figures and Tables
Figure 2
Diffusion weighted images (lower line) of sporadic Creutzfeldt-Jakob disease show asymmetrical high signal intensities along the gyrus, which were not so clear in T2W images (upper line)
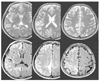
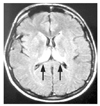
Figure 3
Periodic sharp waves at about 1.5/sec; diffuse, bilateral, slightly asymmetrical: and maximal over the anterior portions of the brain


References
1. Kong Q, Huang S, Zou W, Vanegas D, Wang M, Wu D. Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci. 2005. 25:7944–7949.

2. Marsh RF, Kincaid AE, Bessen RA, Bartz JC. Interspecies transmission of chronic wasting disease prions to squirrel monkeys (Saimiri sciureus). J Virol. 2005. 79:13794–13796.
3. Global surveillance, diagnosis and therapy of human transmissible spongiform encephalopathies: report of a WHO consultation. 9-11 February 1998; Geneva, Switzerland.
4. Cosseddu GM, Agrimi U, Pinto J, Schuddel AA. Advances in scrapie research. Rev Sci Tech. 2007. 26:657–668.
5. Conti S, Masocco M, Toccaceli V, Vichi M, Ladogana A, Almonti S, Puopolo M, Pocchiari M. Mortality from human transmissible spongiform encephalopathies: a record linkage study. Neuropidemiolog. 2005. 24:214–220.
6. Kristiansen M, Deriziotis P, Dimcheff DE, Jackson GS, Ovaa H, Naumann H, Clarke AR, van Leeuwen FWB, Menendez-Benito V, Dantuma NP, Portis JL, Collinge J, Tabrizi SJ. Disease-associated prion protein oligomers inhibit the 26S porteasome. Molecular Cell. 2007. 26:175–187.
7. Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, Alxhekhlee A, Castellani R, Cohen M, Barria M, Gonzalez-Romero D, Belay ED, Schonberger LB, Marder K, Harris C, Burke JR, Montine T, Wisniewski T, Dickson DW, Soto C, Hulette CM, Mastrianni JA, Kong Q, Zou WQ. A Novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 2008. 63:697–708.
8. Prusiner SB. Novel Proteinaceous Infectious Particles Cause Scrapie. Science. 1982. 216:136–144.
9. Riek Roland, Hornemann Simone, Wider Gerhard, Billeter Martin, Glockshuber Rudi, Wüthrich Kurt. NMR structure of the mouse prion protein domain PrP(121~231). Nature. 1996. 382:180–182.
10. Bernoulli C, Siegfried J, Baumgartner G. Danger of accidental person-to-person transmission of Creutzfeldt-Jacob disease by surgery. Lancet. 1977. 1:478–479.
11. Prusiner SB. Prions. Proc Natl Acad Sci. 1998. 95:13363–13383.
12. Chsea R, Piccardo P, Quaglio E. Molecular distinction between pathogenic and infectious properties of the prion protein. J Virol. 2003. 77:7611–7622.
13. Ross Eric D., Edskes Herman K., Terry Michael J., Wickner Reed B.. Primary sequence independence for prion formation. Proc Natl Acad Sci U S A. 2005. 102:12825–12830.
14. Johnson RT, Gibbs CJ Jr. Creutzfeldt-Jakob disease and related transmissible spongiform encephalopathies. N Engl J Med. 1998. 339:1994–2004.
15. Zou WQ, Capellari S, Parchi P, Sy MS, Gambetti P, Chen SG. Identification of novel proteinase K-resistant C-terminal fragments of PrP in Creutzfeldt-Jakob Disease. J Biol Chem. 2003. 278:40429–40436.
16. Parkin ET, Watt NT, Hussain I, Eckman EA, Eckman CB, Manson JC, Baybutt HN, Turner AJ, Hooper NM. Cellular prion protein regulates beta-secretase cleavage of the Alzheimer's amyloid precursor protein. Proc Natl Acad Sci USA. 2007. 104:11062–11067.
17. Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001. 411:810–813.
18. Wells GAH, Scott AC, Johnson CT. A novel progressive spongiform encephalopathy in cattle. Vet Rec. 1987. 121:419–420.
19. Collins SJ, Lawson VA, Masters CL. Transmissible spongiform encephalopathies. Lancet. 2004. 363:51–61.
20. Bruce ME, Will RG, Ironside JW. Transmissions to mice indicate that 'new variant' CJD is caused by the BSE agent. Nature. 1997. 389:498–501.
21. Scott MR, Perezt D, Hguyen HO, Dearmond SJ, Prusiner SB. Transmissions barriers for bovine, ovine and human prions in transgenic mice. J Virol. 2005. 79:5259–5271.
22. Brown P, Will RG, Bradley R, Asher DM, Detwiler L. Bovine spongiform encephalopathy and Creutzfeldt-Jakob disease: background, evolution, and current concerns. Emerg Infect Dis. 2001. 7:6–16.
23. Williams ES, Young S. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildlife Dis. 1980. 16:89–98.
24. Belay ED, Maddox RA, Williams ES, Miller MW, Gambetti P, Schonberger LB. Chronic wasting disease and potential transmission to humans. Emerg Infect Dis. 2004. 10:977–984.
25. Mawhinney S, Pape WJ, Forster JE, Anderson CA, Bosque P, Miller MW. Human prion disease and relative risk associated with chronic wasting disease. Emerg Infect Dis. 2006. 12:1527–1535.
26. Sohn HJ, Kim JH, Choi KS, Nah JJ, Joo YS, Jean YH, Ahn SW, Kim OK, Kim DY, Balachandran A. A case of chronic wasting disease in an elk imported to Korea from Canada. J Vet Med Sci. 2002. 64:855–858.
27. Cali I, Castellani R, Yuan J, Al-Shekhlee A, Cohen ML, Xiao X, Moleres FJ, Parchi P, Zou WQ, Gambetti P. Classification of sporadic Creutzfeldt-Jakob disease revisited. Brain. 2006. 129:2266–2277.
28. Cheong HK, et al. Report No. 01-PJ1-PG3-21900-0010. Epidemiological study on the establishment of surveillance system on Creutzfeldt-Jakob disease. 2003. 05. Ministry of Health and Welfare;(Korean).
29. Korea Centers for Disease Control and Prevention. Reported cases of Creutzfelt-Jacob Disease by year. cited 2008 July 10. Available from: http://sentinel.cdc.go.kr/Cro/Main/Status.asp.
30. Zigas V. Laughing Death, the untold story of Kuru. 1990. New Jersey: Humana Press;197–228.
31. Sikorska B, Liberski PP, Sobow T, Budka H, Ironside JW. Ultrastructural study of florid plaques in variant Creutzfeldt-Jakob disease: a comparison with amyloid plaques in kuru, sporadic Creutzfeldt-Jakob disease and Gerstmann-Sträussler-Scheinker disease. Neuropathol Appl Neurobiol. 2008. 05. 30. (online only).
32. Ladogana A, Puopolo M, Croes EA, Budka H, Jarius C, Collins S, Klug GM, Sutcliffe T, Giulivi A, Alperovitch A, Delasnerie-Laupretre N, Brandel JP, Poser S, Kretzschmar H, Rietveld I, Mitrova E, Cuesta Jde P, Martinez-Martin P, Glatzel M, Aguzzi A, Knight R, Ward H, Pocchiari M, van Duijn CM, Will RG, Zerr I. Mortality from Creutzfeldt-Jakob disease and relaed disorders in Europe, Australia, and Canada. Neurology. 2005. 64:1586–1591.
33. Total cases of CJD: definite and probable cases: sporadic, familial/genetic, vCJD and iatrogenic deaths. EUROCJD. viewed on July 10, 2008. http://www.eurocjd.ed.ac.uk/totalcasesneuro.htm
.
34. National CJD Surveillance Unit. Fifteenth Annual Report 2006: Creutzfeldt-Jakob disease surveillance in the UK. 2007. Edinburgh: National CJD Surveillance Unit.
35. National Prion Disease Pathology Surveillance Center cases examined. National Prion Disease Pathology Surveillance Center (NPDPSC). viewed on July 10, 2008. http://www.cjdsurveillance.com/resources-casereport.html.
36. Brown P, Gibbs CJ Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC. Human spongiform encephalopathy: the National Institutes of Health Series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994. 35:513–529.
37. Bernoulli CC, Masters CL, Gajdusek DC, Gibbs CJ Jr, Harris JO. Prusiner SB, Hadlow WJ, editors. Early clinical features of Creutzfeldt-Jakob disease (subacute spongiform encephalopathy). Slow transmissible diseases of the nervous system, vol 1: clinical, epidemiological, genetic and pathological aspects of the spongiform encephalopathies. 1979. New York: Academic Press;229–241.
38. Brown P, Gibbs CJ Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC. Human spongiform encephalopathy: the National Institutes of Health Series of 300 cases of experimentally transmitted disease. Ann Neurol. 994. 35:513–529.
39. Uro-Coste E, Cassard H, Simon S, Lugan S, Bilheude JM, Perret-Liaudet A, Ironside JW, Haik S, Basset-Leobon C, Lacroux C, Peioch K, Streichenberger N, Langeveld J, Head MW, Grassi J, Hauw JJ, Schelcher F, Delisle MB, Andreoletti O. Beyond PrPres type 1/type 2 dichotomy in Creutzfeldt-Jakob disease. PLoS Pathogens. 2008. 4(e 1000029):1–9. (online onle; www.plospathogens.org).
40. Ludewigs H, Zuber C, Vana K, Nikles D, Zerr I, Weiss S. Therapeutic approaches for prion disorders. Expert Rev Anti Infect Ther. 2007. 5:613–630.
41. Capellari S, Cardone F, Notari S, Schininá ME, Maras B, Sitá D, Baruzzi A, Pocchiari M, Parchi P. Creutzfeldt-Jakob disease associated with the R208H mutation in the prion protein gene. Neurology. 2005. 64:905–907.
42. Mastrianni JA. Power C, Johnson RT, editors. Prion diseases and dementia. Emerging neurological infections. 2005. Boca Raton: Taylor and Francis;77–113.
43. Masters CL, Gajdusek DC, Gibbs CJ Jr. Creutzfeldt-Jakob disease virus isolations from the Gerstmann-Straussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virus - induced spongiform encephalopathies. Brain. 1981. 104:559–588.
44. Max DT. The family that couldn't sleep: a medical mystery. Random hous.
45. Montagna P, Gambetti P, Cortelli P, Lugaresi E. Familial and sporadic fatal insomnia. Lancet Neurol. 2003. 2:167–176.
46. Mastrianni JA. Power C, Johnson RT, editors. Prion diseases and dementia. Emerging neurological infections. 2005. Boca Raton: Taylor and Francis;77–113.
47. Duffy P, Wolf J, Collins G, DeVoe AG, Streeten B, Cowen D. Possible person to person transmission of Creutzfeldt-Jakob disease. N Engl J Med. 1974. 290:692–693.
48. Brown P, Preece M, Brandel JP, Sato T, McShane L, Zerr I, Fletcher A, Will RG, Pocchiari M, Cashman NR, d'Aignaux JH, Cervenakova L, Fradkin J, Schonberger LB, Collins SJ. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology. 200. 55:1075–1081.
49. Zou S, Fang CT, Schonberger LB. Transfusion transmission of human prion disease. Transfusion Medicine rev. 2008. 22:58–69.
50. Walker JT, Dickinson J, Sutton JM, Marsh PD, Raven NDH. Implications for Creutzfeldt-Jakob disease in Dentistry: a Review of Current Knowledge. J Dent Res. 2008. 87:511–519.
51. The National Creutzfeldt-Jakob Surveillance Unit. http://www.cjd.ed.ac.uk/figures.htm.
52. Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith PG. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996. 347:921–925.
53. Will RG, Zeidler M, Stewart GE. Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol. 2000. 47:575–582.
54. Will RG, Zeidler M, Stewart GE, Macleod MA, Ironside JW, Cousens SN, Mackenzie J, Estibeiro K, Green AJ, Knight RS. Diagnosis of new variant Creutzfeldt -Jakob disease. Ann Neurol. 2000. 47:575–582.
55. Peden AH, Ironside JW. Review: pathology of variant Creutzfeldt-Jakob disease. Folio Neuropathol. 2004. 42S:85–91.
56. The National CJD Surveillance Unit. Department of Infectious and Tropical Diseases. London School of Hygiene and Tropical Medicine. Creutzfeldt-Jakob disease surveillance in the UK. Ninth Annual Report. 2000. London, United Kingdom:
57. Jeong BH, Lee KH, Kim NH, Jin JK, Kim JI, Carp RI, Kim YS. Association of sporadic Creutzfeldt-Jakob disease with homozygous genotypes at PRNP codons 129 and 219 in the Korean population. Neurogenetics. 2005. 6:229–232.
58. Collinge J, Whitfield J, McKintosh E, Beck J, Mead S, Thomas DJ, Alpers MP. Kuru in the 21st century-an acquired human prion disease with very long incubation periods. Lancet. 2006. 367:2068–2074.
59. Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. Transmissions to mice indicate that 'new variant' CJD is caused by the BSE agent. Nature. 1997. 389:498–501.
60. Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J, Doey LJ, Lantos P. The same prion strain causes vCJD and BSE. Nature. 1997. 389:448–450.
61. Johnson R. Prion diseases. Lancet Neurol. 2005. 4:635–642.
62. Seitz R, von Auer F, Blümel J, Burger R, Buschmann A, Dietz K, Heiden M, Hitzler WE, Klamm H, Kreil T, Kretzschmar H, Nübling M, Offergeld R, Pauli G, Schottstedt V, Volkers P, Zerr I. Impact of vCJD on blood supply. Biologicals. 2007. 35:79–97.
63. Risk assessment for transmission of vCJD via surgical instruments: a modelling approach and numerical scenarios Summary report. 2001. 03. Economics and Operational research division-Department of Health.
64. Dunstan RA, Alpers MP. Variant Creutzfeldt-Jakob disease: implications for the health care system. Aust NZ J Public Health. 2005. 29:308–312.
65. Wadsworth JD, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Collinge J. Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet. 2001. 358:171–180.
 XML Download
XML Download