

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Introduction
Although oxygen is required for aerobic life, it may be toxic under certain conditions [1,25,37]. Oxidative stress has been shown to play an important role in the pathogenesis of embryo development [4,28,39]. Reactive oxygen species (ROS) are forms of oxygen that result from incomplete reduction of molecular oxygen that may induce different types of cell injury, in particular, lipid peroxidation and membrane damage. Although low levels of ROS play an important role in physiological functions, several studies have reported that high concentrations of ROS are cytotoxic and have been implicated in the pathological conditions such as carcinogenesis of various types of malignancies [3,52,55]. The major oxygen species responsible for these oxidative stresses are hydrogen peroxide (H2O2), the free radical superoxide anion (O2-) and the hydroxyl radical (OH-) [6]. Among the ROS, H2O2 has been implicated as a cellular toxin [19,61]. Since H2O2 has remarkable membrane permeability [23], intracellular H2O2 can induce detrimental effects in cells [20,41,42,49]. In addition, oxidative stress on tissues and cells has been widely recognized as a key factor effecting the development of embryos [13,31,32]. Therefore, the inhibition of such free radical-mediated pathology has become a central focus of research efforts targeted at the prevention or amelioration of embryo or embryonic stem (ES) cell injury. However, the effects of dihydrotestosterone (DHT) on ES cells, in the context of oxidative stress, have not been thoroughly evaluated to date.
DHT is a biologically active metabolite of the hormone testosterone; it is formed primarily in the prostate gland, testes, hair follicles, and adrenal glands by the enzyme 5α-reductase by reduction of the delta-4,5 double-bond of testosterone [15,24,52]. DHT has been shown to be a powerful antioxidant, effectively preventing the formation of lipid peroxides (LPO) [8]. The chemical structure of DHT allows for the donation of an H+ atom to a peroxyl radical [45,60], which results in free radical scavenging and may exert additional effects by early interference or during the propagation phase of LPO. It is postulated that this antioxidant activity is one mechanism by which DHT confers cardio-, hepato- and, neuro- protection. Despite their antioxidant activities, it is not known whether DHT prevents or alleviates the ES cell toxicity induced by oxidative stress.
ES cell lines are pluripotent cells derived from the blastocyst-stage of mammalian embryos [12,16]. These unique cells are characterized by their capacity for prolonged undifferentiated proliferation in culture with the potential to differentiate into derivatives of all three germ layers [46,47,51]. In addition, these cells closely resemble their in vivo counterparts, providing a stable in vitro model of embryo growth and development that acts as a model for the study of specific cell signaling systems [30,33]. Therefore, the study of mouse ES cells has provided a versatile biological system that has led to major advances in cell and developmental biology. Many studies have used H2O2 to investigate the mechanisms of cell injury resulting from acute oxidative stress in various cells and tissues [18,59]. Therefore, the purpose of this study was to determine the effect of DHT on mouse ES cells under conditions of H2O2-induced oxidative stress.
Materials and Methods
Materials
The mouse ES cells were obtained from the American Type Culture Collection (ES-E14TG2a). The fetal bovine serum (FBS) was purchased from Gibco (USA). DHT was acquired from TCI (Tokyo Kasei Kogyo, Japan). The 5-(and-6)-chloromethyl-2',7'-dichlorodihydrofluorescein diacetate acetyl ester (DCF-DA) was purchased from Molecular Probes (USA). Hydrogen peroxide solutions were obtained from Sigma-Aldrich (USA). Phospho-p38 mitogen activated protein kinase (MAPK), p38 MAPK, phospho-c-Jun N-terminal kinase/stress activated protein kinase (SAPK/JNK), SAPK/JNK, and phospho-nuclear factor (NF)-κB, and NF-κB antibodies were purchased from New England Biolabs (UK). Cyclin D1, cyclin E, cyclin dependent protein kinase (CDK) 2, and CDK 4 antibodies were purchased from Santa Cruz Biotechnology (USA). Goat anti-rabbit IgG were obtained from Jackson Immunoresearch (USA). Liquiscint was obtained from National Diagnostics (USA).
ES cell culture
Mouse ES cells were cultured for 5 days in Dulbecco's Modified Eagle Media (DMEM; Gibco-BRL, USA) supplemented with 3.7 g/l sodium bicarbonate, 1% penicillin and streptomycin, 1.7 mM L-glutamine, 0.1 mM β-mercaptoethanol, 5 ng/ml mouse leukemia inhibitory factor and 15% FBS. The cells were grown on either gelatinized 12-well plates or a 60 mm culture dish in an incubator maintained at 37℃ in an atmosphere containing 5% CO2 and air.
Assay of intracellular reactive oxygen species
The cells were pretreated with or without DHT for 30 min and then were treated with H2O2 for 0 to 120 min. DCF-DA was used for the detection of H2O2-induced ROS, which acts as an H2O2-sensitive fluorophore. Next, 10 µM DCF-DA was added to the cells, which were then incubated in the dark for 30 min at room temperature. The cells were then viewed using a laser confocal microscope (×400; fluoview 300; Olympus, Japan), fluorescence was excited at 488 nm and emitted light was observed at 515-540 nm.
To quantify the intracellular H2O2 levels, the cells treated with DCF-DA were rinsed twice with ice-cold PBS then scraped. A 100 µl cell suspension was loaded into a 96-well plate and examined using a luminometer (Victor3; PerkinElmer, Finland) and a fluorescent plate reader with excitation and emission wavelengths of 485 nm and 535 nm, respectively.
DNA fragmentation assay
The cells were pretreated with or without DHT for 30 min and then were treated with H2O2 for 24 h. The cells were then suspended in lysis buffer (10 mmol/l Tris-HCl pH 7.5, 10 mmol/l EDTA pH 8.0, 0.5% Triton-X). The cell lysates were treated with 200 µg/ml proteinase K at 60℃ for 6 h followed by DNA extraction using phenol-chloroform. The extracted DNA was precipitated using isopropyl alcohol, then digested with 10 µg/ml TE-RNase at 37℃ for 1 h. After digestion, the product was electrophoresed on a 1% agarose gel stained with ethidium bromide and photographed under ultraviolet light.
Measurement of lipid peroxides
The cells were pretreated with or without DHT for 30 min and then were treated with H2O2 for 0 to 120 min. The level of LPO in the monolayer cells was determined by measuring the malondealdehyde content according to the description of Ohkawa et al. [48]. The mouse ES cells were washed twice with PBS, then harvested and sonicated. One hundred microliters of sonicated cells were mixed with 8% SDS (100 µl), 0.8% 2-thiobarbituric acid (TBA; 200 µl) and 20% acetic acid (200 µl). The mixture was heated to 95℃ for 60 min, then cooled in ice water. To extract nonspecific red pigment, a n-butanol-pyridine mixture (15 : 1 vol/vol, 1 ml) was added, then shaken vigorously and centrifuged at 4,000 rpm for 10 min. The upper organic layer was measured using spectrofluorometry at an emission wavelength of 553 nm with an excitation wavelength of 515 nm. The 1,1,3,3-tetraethoxypropane was used as a standard, and the values of LPO for the samples were expressed as nmol/mg protein. Taurine was added to cell mixtures to prevent any initiation of membrane lipid peroxidation during the assay. An addition of taurine to the standard 1,1,3,3-tetraethoxypropane or a control sample did not affect the color development with the TBA (data not shown).
Trypan blue exclusion assay
The cells were pretreated with or without DHT for 30 min and then were treated with H2O2 for 24 h and then washed twice with PBS. The cells were then detached from the culture dishes using a 0.05% trypsin and 0.5 mM EDTA solution, and this action was quenched with a soybean trypsin inhibitor (0.05 mg/ml). Subsequently, a 0.4% (w/v) trypan blue solution (500 µl) was added to the cell suspension and the cells were counted using a hemocytometer while keeping a separate count of the blue cells. The cells that failed to exclude the dye were considered non-viable; therefore, the data is expressed as a percentage of viable cells. Cell injury was assessed by measuring the LDH activity in the medium using a LDH assay kit (Iatron Lab, Japan). The level of LDH released is expressed as a percentage of the control value.
[3H] thymidine incorporation
The [3H] thymidine incorporation experiments were carried out as described by Brett et al. [11]. In this study, the cells were cultured in one well until they reached 50% confluence, at which time they were washed twice with PBS, then maintained in serum-free DMEM with all supplements and incubated for 24 h. The cells were pretreated with or without DHT for 30 min and then were treated with H2O2 for 24 h. Next, 1 µCi of [methyl-3H] thymidine (specific activity: 74 GBq/mmol, 2.0 Ci/mmol; Amersham Biosciences, UK) was added to the cultures and the incubation continued for 1 h at 37℃. The cells were then washed twice with PBS, fixed in 10% trichloroacetic acid (TCA) at 23℃ for 15 min, then washed twice with 5% TCA. The acid-insoluble material was dissolved in 0.2 N NaOH for 12 h at 23℃. Aliquots were removed and their radioactivity determined using a liquid scintillation counter (LS 6500; Beckman, USA). All values are means ± SE of triplicate experiments. Values were converted from absolute counts to a percentage of the control to allow for comparison between experiments.
Western blot analysis
The cell homogenates (20 µg protein) were separated by electrophoresis on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. After the blots were washed with TBST [10 mM Tris-HCl (pH 7.6), 150 mM NaCl, 0.05% Tween-20], the membranes were blocked with 5% skim milk for 1 h and incubated with the appropriate primary antibodies at the dilutions recommended by the supplier. The membrane was then washed and the primary antibodies detected using goat anti-rabbit IgG or goat anti-mouse IgG conjugated to horseradish peroxidase. The bands were visualized using enhanced chemiluminescence (Amersham Pharmacia Biotech, UK).
Results
Effect of DHT on H2O2-induced cell injury
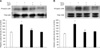
We determined the H2O2-induced ROS generation in mouse embryonic stem cells using DCF-DA (10-5 M), a fluorescent probe of intracellular H2O2. Production of ROS was detected in the cells exposed to H2O2 10-3 M for a variety of times (0-60 min) by the DCF fluorescence intensity in the ES cells. As shown Figs. 1A and B the level of ROS generation was increased after ≥ 10 min. In addition, H2O2 treatment significantly increased LPO formation after ≥ 30 min compared with controls as shown in Fig. 1C. In order to examine the effect of H2O2 on cell injury, the ES cells were incubated for a variety of times (0-24 h) with H2O2 exposure and the level of DNA fragmentation was examined. The H2O2 increased the level of DNA fragmentation after ≥ 16 h (Fig. 1D). However, the H2O2-induced cell injury was prevented by DHT (10-7 M) pretreatment. As shown in Figs. 2A and B increase in the ROS generation and LPO formation by H2O2 were inhibited by DHT. In addition, as shown in Figs. 2C and D the increase of LDH release and the decrease of cell viability, with H2O2, were inhibited by DHT. This is further supported by the assessment of the DNA fragmentation experiments (Fig. 2E).
Effect of DHT on H2O2-induced activation of p38 MAPKs, SAPK/JNK and NF-κB
In order to assess the involvement of MAPKs in H2O2-induced cell injury, the cells were treated with H2O2 for 30 min and then phosphorylation of MAPKs was measured by Western blot analysis. As shown in Figs. 3A and B, H2O2 increased p38 MAPK and SAPK/JNK phosphorylation, which was inhibited by DHT. In the next step, the involvement of NF-κB activation in H2O2-induced cell injury was evaluated. As shown in Fig. 4A, H2O2-induced NF-κB activation was inhibited by DHT treatment. In addition, NF-κB phosphorylation was attenuated by pretreatment with SB 203580 (a p38 MAPK inhibitor) and SP 600125 (a SAPK/JNK inhibitor, 10-6 M) (Fig. 4B).
Effect of DHT on H2O2-induced DNA synthesis regulation
To investigate the effect of DHT on H2O2-induced increase of cell cycle regulatory protein expression, the mouse ES cells were pretreated with DHT for 30 min prior to adding the H2O2. The H2O2-induced decrease of cell cycle regulatory protein (cyclin D1, cyclin E, CDK 2, and CDK 4) expression levels were recovered by pretreatment with DHT in the mouse ES cells (Figs. 5A, B, C and D). In addition, as shown in Fig. 5E, the maximum decrease in the level of [3H] thymidine incorporation was observed following 24 h of H2O2 exposure (30% decrease vs. Control; *p < 0.05). However, this inhibitory effect of H2O2 on DNA synthesis were blocked by pretreatment with DHT, SB 203580 (p38 MAPK inhibitor, 10-6 M), SP 600125 (SAPK/JNK inhibitor, 10-6 M), SN 50 (NF-κB phosphorylation inhibitor, 10-6 M), and Vitamin C (antioxidant, 10-3 M).
Discussion
In the present study, we showed that DHT prevented H2O2-induced cell injury likely by the downregulation of p38 MAPK, SAPK/JNK and NF-κB in mouse ES cells. Oxidative stress induced by ROS is a well known cause of several physiological and pathophysiological processes including cell proliferation and differentiation [26,50]. ROS, which include the superoxide anion (O2-), H2O2, OH, and singlet oxygen (1O2) are formed by incomplete reduction of molecular oxygen. ROS may cause different types of cell injury, particularly lipid peroxidation and membrane damage [6]. H2O2 is often used as a model compound to induce oxidative stress in cell systems [54]. The antioxidant properties of DHT have not been fully studied and multiple mechanisms may be involved in its action [10,21,22]. LPO formation in biological membranes is a free radical-mediated event and is regulated by the availability of substrates in the form of polyunsaturated fatty acids, pro-oxidants which promote peroxidation, and inhibited by antioxidants such as β-carotene and superoxide dismutase [14,27,55]. Elevated levels of LPO have been linked to cell injury such as the increase in permeability to ions and eventually the disruption of the cell membrane leading to release of cell organelles [14,27,55]. The results of this study showed that exposure to 1 mM H2O2 generated LPO, and DHT attenuated the LPO formation. The findings of this study demonstrated that the protective effects of DHT were associated with the inhibition of LPO generation, which is consistent with the results of previous reports [40,62]. Bennett et al. [7] reported that testosterone with a keto group at the C3 position, showed a mild preventive effect compared to DHT. Thus we suggest that the protective effects of DHT are due to its basic chemical properties as a hydrophobic phenolic molecule.
H2O2-induced oxidative stress mediates the phosphorylation of protein kinases through a series of signal cascades, such as activation of MAPKs and, a nuclear factor-κB [44]. The MAPKs proteins are mediators of signal transduction from the cell surface to the nucleus and play a major role in triggering and coordinating gene responses [36]. In addition, phosphorylation of JNK and p38 MAPK has been shown to play a role in cellular differentiation and inflammatory responses [17,29,35]. Antioxidants have been shown to attenuate the activation of MAPKs signaling [29,58], thereby indicating that the MAPK signaling pathway is an important target of ROS. The levels of intracellular ROS were increased as the result of the addition of extracellular H2O2, which subsequently increased p38 MAPK and SAPK/JNK phosphorylation. In the present study, DHT treatment was observed to inhibit the effects of H2O2 on MAPKs phosphorylation, which supports the role of DHT in this process. In addition, H2O2 activated NF-κB is one of the key regulatory molecules in oxidative stress-induced cell injury and the activation of NF-κB has an important regulatory role in cell proliferation and oncogenesis [2,43,56]. NF-κB is commonly activated by oxidants, including H2O2 [34]. In this study, we demonstrated that NF-κB was activated by H2O2 treatment. In addition, activated NF-κB was blocked by SB 203580 (p38 MAPK inhibitor) and SP 600125 (SAPK/JNK inhibitor). These results suggest that NF-κB is a down stream target of JNK and p38 MAPK. In addition, DHT treatment inhibited the H2O2-induced activation of these signaling molecules. Moreover, the p38 protein of the MAPK family has been shown to modulate NF-κB activation [9,57]. The inhibition of H2O2-induced phosporylation of MAPKs proteins may result in the prevention of downstream events such as activation of NF-κB [44]. Therefore, we suggest that the significant inhibition of H2O2-induced phosphorylation of MAPKs by DHT might be responsible for the inhibitory effects on the activation of the transcription factor NF-κB.
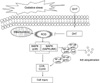
Cyclin-dependent kinases (CDK 2 and CDK 4) are enzymes responsible for the coordinated progression of the cell cycle [53]. CDKs are activated by their association with the D-type cyclins (cyclin D1, D2 and D3); cyclin D1 plays an important role in the G1 phase of cell cycle progression in association with CDK 4 [53]. H2O2 is known to induce cell cycle arrest and apoptosis in various cell types cultured in vitro [5]. MAPKs also lead to the increase of cell cycle regulatory proteins [cyclin D1, cyclin E, CDK 2, and CDK 4] [38]. In this study, H2O2 decreased the expression of cell cycle regulatory proteins. However, DHT inhibited the effect of H2O2-induced decrease of cell cycle regulatory protein expression. Moreover, H2O2 decreased DNA synthesis. However, DHT inhibited the effect of the H2O2-induced decrease of DNA synthesis. Moreover, the H2O2 decrease of DNA synthesis was not only inhibited by DHT treatment but also other antioxidants (vitamin C), SB 203580, SP 600125, and SN 50. These findings suggest that H2O2-induced activation of p38 MAPK, SAPK/JNK and NF-κB are involved in the mechanism associated with decreased DNA synthesis (Fig. 6).
In conclusion, the results of this study confirmed the protective effects of DHT were associated with the inhibition of p38 MAPK, SAPK/JNK and NF-κB signaling pathways by free radical scavenging. These data demonstrate that DHT appears to have powerful cytoprotective activity against H2O2-induced ES cell injury.




 XML Download
XML Download