

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
In mammalian tissues, reactive oxygen species (ROS), such as superoxide radicals (O2-), hydroxyl radicals (OH-) and hydrogen peroxide (H2O2), are continuously generated during aerobic metabolism. ROS modulate multiple cellular processes, including proliferation, differentiation, and signaling. However, excessive generation of ROS can cause detrimental changes, such as lipid peroxidation, DNA breakage, protein degradation, and enzyme inactivation [6,26].
The superoxide dismutase (SOD) family is a ubiquitously distributed group of enzymes that efficiently catalyze the dismutation of O2- [15]. The cytoplasmic Cu/Zn SOD (SOD1) is located in multiple intracellular compartments including the cytosol, nucleus, lysosome, and mitochondrial intermembrane space [4,20], while the manganese SOD (SOD2) is only located in mitochondria [28].
Mice lacking SOD1 develop a range of pathologies, including hepatocellular carcinoma, cochlear hair cell loss, vascular dysfunction, anemia, acceleration of age-related loss of muscle mass, an earlier incidence of cataracts, and a reduced lifespan [7,8,13,16,17,23]. Pregnant female mice lacking SOD1 experienced an increased incidence of lethality in their embryos [11]. Co-treatment with SOD alleviated the teratogenicity of a broad range of embryonic cytotoxic agents, including ethanol and hyperglycemia [3,29]. SOD1 mRNA was detected in all embryos at embryonic day (ED) 7.5-18.5 and was expressed in a cell- and tissue-specific manner in developing organs [30]. In this study, we investigated the expression patterns of SOD1 protein during embryonic development, using Western blotting and immunohistochemical analysis, to better understand the function of SOD1 in embryogenesis.
Materials and Methods
Experimental animals
Male and female ICR mice (8 to 10 weeks old) were purchased from a commercial breeder, Biogenomics (Korea). All animals were housed in polycarbonate cages and acclimatized for 1 wk. The environmental conditions were controlled, with an ambient temperature of 21 ± 2℃, relative humidity of 55% ± 10%, air ventilation rate of 10 cycles per h, and a 12 : 12 h light : dark cycle. The animals were fed a standard mouse chow (Samyang, Korea) and tap water ad libitum throughout the experimental period. All experiments were approved and performed according to the "Guide for Care and Use of Animals" (Chungbuk National University Animal Care Committee, Korea).
Preparation of embryos and extraembryonic tissues
One male and two female mice were housed in cages for mating. Successful mating was confirmed by the presence of a copulation plug in the vagina. The day on which the vaginal plug was observed was designated ED 0.5. Pregnant mice were sacrificed under pentobarbital anesthesia on ED 8.5-18.5, and embryos and extraembryonic tissues, including the placenta, were dissected.
Protein extraction and Western blotting analysis
Total protein was extracted from mouse embryos and extraembryonic tissues taken on ED 8.5-18.5. Tissue samples were homogenized in lysis buffer (50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 1 µg/ml aprotinin, 0.5 mM sodium orthovanadate, and 20 mM sodium pyrophosphate). The lysates were clarified by centrifugation at 14,000 rpm for 10 min and incubated at 95℃ in loading buffer (0.125 M Tris-HCl (pH 6.8), 4% SDS, 20% glycerol, 10% 2-mercaptoethanol, and 0.002% bromophenol blue) for 5 min before electrophoresis. Aliquots containing 60 µg of total protein were separated by SDS-PAGE (using 10% polyacrylamide gels) and transferred to nitrocellulose membranes (Immobilon; Millipore, USA). The membranes were blocked with 5% non-fat milk in Tris-buffered saline [TBS; 25 mM Tris (pH 7.4), 200 mM NaCl] for 1 h at room temperature to eliminate nonspecific binding, and were then incubated with the primary antibodies (diluted 1 : 1,000 in 5% non-fat milk in TBST), polyclonal rabbit anti-SOD1 (Stressgen, Canada) and polyclonal rabbit anti-β-actin (Cell Signaling, USA). Next, membranes were incubated with a horseradish peroxidase-conjugated secondary antibody (1 : 1,000; Cell Signaling, USA) for 1 h at room temperature, visualized using a Western Lightning Chemiluminescence reagent (Perkin-Elmer Life Sciences, USA) according to the manufacturer's protocol, and photographed (Gel Doc EQ; Bio-Rad, USA). Immunoreactive bands were quantified and normalized using PDQuest Image software (Bio-Rad, USA). Experiments were performed in triplicate, and data are presented as mean ± SD.
Immunohistochemistry
For immunohistochemical analysis, mouse embryos obtained on ED 13.5-18.5 were fixed in Bouin solution. Tissue sections were deparaffinized with xylene and rehydrated through an ethanol gradient. Endogenous peroxidase activity was quenched by 0.3% hydrogen peroxide in methanol for 15 min, and then sections were washed 4 times in PBS, for 5 min each time. Nonspecific binding was blocked with a 20 min incubation in diluted normal serum, and sections were incubated for 1 h at 37℃ with the SOD1 antibody. Next, sections were incubated for 30 min at room temperature with a biotinylated secondary antibody (Vector, USA) followed by 40 min at room temperature with the peroxidase-conjugated biotin-avidin complex (Vectastain ABC kit; Vector, USA). Finally, the bound peroxidase was revealed by immersing the sections in diaminobenzidine (Vector, USA). Sections were counterstained with hematoxylin, rehydrated for 15 min in deionized water to remove any precipitated Tris, and then dehydrated in a series of ethanol and xylene. Negative control experiments for the antibodies were performed as above with the omission of the primary antibody.
Results
Expression of SOD1 in developing embryos and extraembryonic tissues
Using Western blot analysis with β-actin as an internal standard, SOD1 was detected in all embryos and extraembryonic tissues from ED 8.5 to 18.5. The signal in embryos was observed at the lowest levels on ED 9.5-11.5 and the highest levels on ED 17.5-18.5 (Fig. 1A). Extraembryonic expression of SOD1 remained constant throughout all stages of embryonic development (Fig. 1B).
Localization pattern of SOD1 during organogenesis
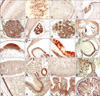
SOD1 immunoreactivity was detected in various embryonic tissues from ED 13.5 to 18.5. The distribution and quantitative expression of SOD1 in developing embryos are described in detail below (Figs. 2 and 3) and are summarized in Table 1. In developing brains, SOD1 immunoreactivity was detected at moderate levels in the external layers (Fig. 2A; asterisk) and pyramidal cells of the cerebral cortex on ED 13.5 to 18.5 (Fig. 2B) and in the cerebellum on ED 17.5 and 18.5 (Fig. 2C). A strong SOD1 signal was observed in the ependymal epithelium of the choroid plexus on ED 13.5 to 18.5 (Fig. 2D) and in Rathke's pouch (hypophysis) after ED 15.5 (Figs. 2E and F). The SOD1 signal was also strong in the developing spinal ganglia on ED 13.5 to 17.5, but was present at only a moderate level on ED 18.5 (Fig. 2G). Moderate SOD1 immunoreactivity was detected in the spinal cord on ED 13.5 to 16.5, and the signal was strong on ED 17.5 and 18.5 (Table 1).
After ED 15.5, SOD1 was strongly expressed in the sensory cells of the cochlear and vestibular epithelia of the inner ear (Figs. 2H and I) and in the olfactory cells and Bowman's glands in the olfactory epithelium (Figs. 2J and K). In developing eyes, strong SOD1 immunoreactivity was observed in the lens fiber and the retinal pigment epithelium (RPE) on ED 16.5, and in the iris and RPE on ED 18.5 (Fig. 2L).
In the parenchyma of the developing lung, weak expression of SOD1 was observed on EDs 13.5 to 15.5, becoming moderate on ED 16.5 to 18.5. Strong SOD1 immunoreactivity was observed in the bronchial and terminal bronchiolar epithelia after ED 15.5 (Figs. 2M and N). SOD1 was weakly expressed in the myocardium of the developing heart up until ED 16.5, and increased slightly after ED 17.5 (Fig. 2O). In contrast, strong SOD1 immunoreactivity was consistently observed in nucleated and enucleated blood cells and in blood vessels (Figs. 2P and Q).
SOD1 was strongly expressed in the basal cells and muscular layers of the stomach on ED 16.5 (Fig. 2R). In developing intestine, SOD1 expression was moderate in the intestinal villi and muscle layers after ED 15.5, but was strong in the goblet cells and basal cell part of the crypt of Lieberkuhn after ED 16.5 (Fig. 2S and T). In the developing liver, strong SOD1 immunoreactivity was observed in hepatocytes and hematopoietic cells during all periods examined (Figs. 3A and B). In the developing pancreas, moderate SOD1 immunoreactivity was observed in the acinus and islet on ED 14.5 to 16.5. After ED 17.5, the SOD1 signal was higher in the acinus than the islet of the pancreas (Fig. 3C). Strong SOD1 immunoreactivity was observed in secretory acini and ducts of the submandibular glands on EDs 17.5 to 18.5 (Fig. 3D).
In the developing kidney, SOD1 immunoreactivity was higher in the cortex than in the medulla, but the signal intensity was moderate in the metanephric corpuscles and tubules on ED 13.5 to 16.5 (Fig. 3E). On ED 17.5, a strong SOD1 signal was observed in the glomeruli, but moderate in the tubules. However, the signal intensity in tubules and glomeruli was reversed on ED 18.5 (Table 1). Moderate SOD1 immunoreactivity was observed in the luminal epithelium of the urinary bladder on EDs 15.5 and 16.5. On ED 18.5, SOD1 immunoreactivity was strong in the surrounding muscular layers, but was weak in the epithelium (Table 1).
In the developing adrenal glands, SOD1 expression was higher in the medulla than in the cortex on EDs 13.5 and 14.5 but was similar in both areas on ED 15.5. On ED 16.5 to 18.5, the SOD1 signal in the cortex had exceeded that observed in the medulla (Figs. 3F and G).
Strong SOD1 expression was observed in the thymus on ED 15.5 to 17.5 (Fig. 3H), whereas expression was moderate on ED 18.5. In the developing lymph nodes, SOD1 expression was weak on ED 14.5 and was high after ED 15.5 (Fig. 3I). In the developing bone and cartilage, a very strong SOD1 signal was observed in chondrocytes on ED 13.5 to 18.5 (Figs. 3J and K). Strong SOD1 immunoreactivity was observed in the surface epithelium, hair follicles, and muscle layers of the skin after ED 15.5 (Figs. 3L and M) and in a collagen fiber-like tissue on ED 18.5 (Fig. 3N).
Discussion
ROS are important in normal developmental processes, such as progression to the early stage, neuronal differentiation, and digit formation. However, excessive production of ROS can cause severe embryonic damage, resulting in embryonic death, preeclampsia, or congenital anomalies in placentation [6,26]. SOD1 plays a major role in antioxidant defense by converting O2- to H2O2 and water [15]. Recently, we demonstrated that SOD1 transcripts were present at all embryonic stages [30]. Here, we confirm the expression pattern of SOD1 protein during normal embryonic development. SOD1 was detected throughout all embryonic stages, in both embryonic and extraembryonic tissues, from ED 8.5 to 18.5. The SOD1 signal was increased at late stages of embryonic development, but maintained a constant level of expression in extraembryonic tissues during organogenesis. SOD1 immunoreactivity was particularly strong in metabolically active sites and in protective tissues, such as the skin and lung, at late gestation.
Uteroplacental circulation is established after implantation. The placenta receives nutrients, oxygen, antibodies, and hormones from the mother's blood and passes out waste [10]. The expression of SOD1 within the human placenta suggests that it might serve an important function at the maternal-fetal interface [27]. The oxygen requirements in rodent embryos vary during embryo development [2]. As the fetus moves from a hypoxic to a relatively hyperoxic environment, accompanying changes in antioxidant enzymes constitute a compensatory mechanism aimed at protecting the newborn from oxidative stress [14].
SOD1 accumulates in many neuronal populations, but it is particularly abundant in motor neurons of the spinal cord [21]. A number of sporadic and familial motor neuron diseases result from point mutations in the gene encoding SOD1 on human chromosome 21 [24]. In this study, SOD1 expression was higher in the spinal cord and spinal ganglia than in the brain during organogenesis.
SOD1 immunoreactivity was ubiquitously present in developing organs and was particularly strong in the ependymal epithelium of the choroid plexus, ganglia, sensory cells of the olfactory and vestibulocochlear epithelia, blood cells and vessels, hepatocytes and hematopoietic cells of livers, lymph nodes, osteogenic tissues, and skin during organogenesis. These results are in agreement with the expression patterns of SOD1 transcripts in most tissues and metabolically active sites [30]. SOD1 levels increase in mature cells to inhibit oxidative stress induced by exogenous materials, such as alcohol, kanamycin, 6-hydroxydopamine, and 1-methyl-4-phenylpyridinium [1,22]. SOD1 immunoreactivity is detected in the epithelial cells of the gastrointestinal tract, respiratory tract, pancreatic islets, and kidneys during late gestation in rats [18,19].
ROS are vital to normal developmental processes, such as proliferation and differentiation [6]. SOD1 is distributed primarily in tissues and cells that are frequently exposed to O2- or its metabolites. In this study, strong SOD1 immunoreactivity was observed in sites of active differentiation and proliferation, such as chondrocytes of osteogenic tissues, hematopoietic cells of the liver, and circulatory blood cells. Erythrocytes use SOD1 to scavenge O2-, but carry only the SOD1 protein, not mRNA, as they lack mitochondria. SOD1 deficiency leads to increased erythrocyte vulnerability, as a consequence of increased oxidation of proteins and lipids. The short lifespan of erythrocytes in SOD1-/- mice has been attributed to both endogenous and environmental factors [12]. Neutrophils produce enormous quantities of O2- through the activity of oxidant-generating systems [25]. High levels of SOD1 activity were detected in cartilage and connective tissues [9], and a low molecular weight mimetic of SOD attenuates chronic inflammation, tissue damage, and bone damage associated with collagen-induced arthritis [5]. Thus, SOD1 may act as an important antioxidant in osteogenesis and hematopoiesis during embryonic development.
SOD1 levels vary widely in different cell types, and even within a particular cell type, during the course of embryonic development. SOD1 may play a crucial role as an antioxidant against ROS in cellular differentiation, regulation, and signaling during murine embryonic development.



 XML Download
XML Download